
嘉峪检测网 2025-02-14 08:36
导读:国家药监局药审中心于2025年02月09日发布了化学药品注册受理审查指南(试行),要求自2025年3月10日起施行,现就本指南要点进行分析如下。
国家药监局药审中心于2025年02月09日发布了化学药品注册受理审查指南(试行),要求自2025年3月10日起施行,现就本指南要点进行分析如下:
第一部分
|
第一部分新旧版对比(2020年 vs 2025年) |
|||
|
对比项 |
2020年版 |
2025年版 |
核心差异说明 |
|
适用范围 |
注册分类1、2、5.1类的临床试验/上市许可申请 |
注册分类1、2、5.1类的临床试验/上市许可申请 |
无变化 |
|
受理部门 |
国家药监局药品审评中心 |
国家药监局药品审评中心 |
无变化 |
|
资料提交形式 |
纸质资料为主,需加盖骑缝章;少量提及电子文件。 |
新增电子化要求: |
新版强化电子化申报流程,适应数字化转型。 |
|
申请表要求 |
需通过报盘程序生成RVT文件,纸质与电子版一致。 |
改为在线填报:通过国家药监局政务服务门户在线提交,强调数据核对码一致。 |
新版改为在线填报。 |
|
形式审查要点 |
包括申报事项、沟通交流、申请表等审查,部分条款较简略。 |
细化条款: |
新版细化条款。 |
|
小微企业优惠 |
需提交纸质证明(如纳税申报表)。 |
简化流程:允许通过电子文件提交证明,减少纸质材料。 |
新版简化流程。 |
|
附件内容 |
自查表包含基础申报信息检查。 |
升级自查表: |
新版自查更全面,覆盖研发到申报全环节。 |
|
特殊药品管理 |
明确麻醉、精神类药品不得委托生产。 |
扩展范围:新增含麻醉/精神药品的复方制剂不得委托生产。 |
新版进一步收紧高风险药品生产管理。 |
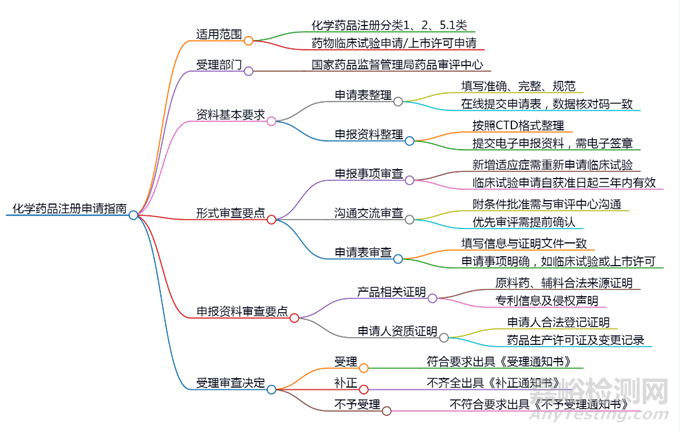
1、适用范围
化学药品注册分类:化学药品注册分类1类、2类、5.1类药物的临床试验申请及上市许可申请。
受理部门:国家药品监督管理局药品审评中心。
2、资料基本要求
申报资料格式:应按照《药品注册管理办法》及《化学药品注册分类及申报资料要求》的规定整理,采用CTD格式。
申请表的整理:
● 填写申请表、申报资料自查表等时需准确、完整、规范,不得手写或涂改。
● 申请人需通过国家药监局政务服务门户填报申请表并在线提交。
3、形式审查要点
(一)申报事项审查要点
1.药物临床试验申请:
● 药物若增加适应症或联合用药,需重新提出申请。
● 临床试验终止后,若需继续进行,需重新申请。
● 不同适应症的药物需分别提交注册申请。
2.非处方药上市许可申请:符合相关情形的可直接提出申请。
3.重大变更:如审评期间发生重大变更,需撤回原申请并重新申报。
(二)沟通交流审查要点
申请附条件批准或优先审评的,需与药品审评中心沟通确认。
(三)申请表审查要点
● 申请表填写要求:
应与证明性文件一致,特别是在药品注册分类、通用名称等信息上。
需说明是否涉及特殊管理药品。
4、申报资料审查要点
(一)产品相关证明性文件
1.原料药、药用辅料及药包材:
● 提供合法来源证明文件包括供货协议、发票等适用于制剂未选用已登记原辅包情形)。
● 提供授权使用书(适用于制剂选用已登记原辅包情形)。如为供应商出具,需有原料药、药用辅料和药包材企业授权,并附授权书。
2.专利信息:
● 需声明不构成侵权,并承诺承担相关责任。
3.临床试验相关证明文件:
● 包括《药物临床试验批件》和临床试验登记号等。
(二)申请人/生产企业证明性文件
● 提供合法登记证明文件及药品生产许可证。
5、其他提示
● 麻醉药品、精神药品:不得委托生产,需遵循相关规定。
● 境外生产药品:提交的证明文件可不经公证。
6、受理审查决定
● 受理通知书:符合要求的出具受理通知书。
● 补正与不予受理:需一次性告知补正内容或不予受理的理由。
7、附件
包含化学药品注册申报资料自查表和参考目录。
第二部分
|
第二部分新旧版对比(2020年 vs 2025年) |
|||
|
对比项 |
2020 年版 |
2025 年版 |
核心差异说明 |
|
适用范围 |
注册分类 3、4、5.2 类的临床试验/上市许可申请 |
注册分类 3、4、5.2 类的临床试验/上市许可申请 |
无变化 |
|
受理部门 |
国家药监局药品审评中心 |
国家药监局药品审评中心 |
无变化 |
|
资料提交形式 |
纸质资料为主,需加盖公章 |
新增电子申报要求:支持 eCTD 格式、电子签章、网络传输提交 |
新版强化电子化申报流程,适应数字化转型。 |
|
专利声明要求 |
需提交专利不侵权声明 |
细化要求:需对照中国上市药品专利信息登记平台声明,遵循《药品专利纠纷早期解决机制》 |
新版加强专利合规管理,明确具体操作流程。 |
|
参比制剂信息 |
需填写参比制剂目录信息 |
新增要求:需明确参比制剂的来源、批准信息、购买凭证等细节 |
新版对参比制剂的真实性核查更严格。 |
|
临床试验安全信息 |
未明确要求 |
新增条目:需提交临床试验进展、安全性更新报告(DSUR)、快速报告安全信息(SUSAR)等 |
新版强化临床试验全流程风险管理。 |
|
境外文件认证 |
需经公证及驻外使领馆认证 |
简化流程:符合 WHO 格式或《取消外国公文书认证要求的公约》的文件可免公证 |
新版优化国际文件认证流程,减轻企业负担。 |
|
自查表内容 |
包含基本资料合规性自查 |
扩展条目:新增生产工艺验证报告、电子签章合规性、临床试验安全信息评估等 |
新版自查更全面,覆盖研发到申报全环节。 |
|
委托生产限制 |
麻醉、精神类药品不得委托生产 |
新增限制:含麻醉、精神类成分的复方制剂也不得委托生产 |
新版进一步收紧高风险药品生产管理。 |
|
药械组合产品申报 |
需提交属性界定结果通知书 |
细化要求:医疗器械部分需单独准备光盘提交 |
新版明确药械组合产品的分类申报细节。 |
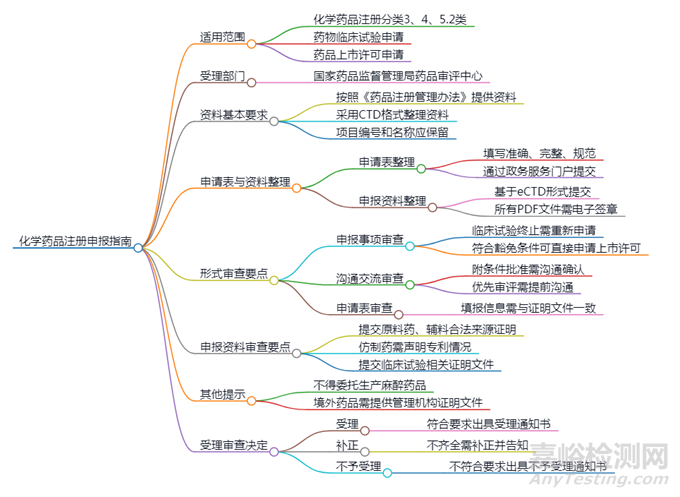
1、适用范围
● 化学药品注册分类:3类、4类和5.2类药物的临床试验申请和上市许可申请。
● 受理部门:国家药品监督管理局药品审评中心。
2、资料基本要求
● 申报资料需遵循《药品注册管理办法》和《化学药品注册分类及申报资料要求》的规定。
● 资料应按照《M4:人用药物注册申请通用技术文档(CTD)》格式整理,确保目录及项目编号不变。
3、申请表和申报资料整理
1.申请表的整理:
● 填写药品注册申请表和自查表时需准确、完整、规范,不得手写或涂改。
● 申请人需通过国家药品监督管理局政务服务门户在线提交申请表。
2.申报资料的整理:
● 提交电子通用技术文档(eCTD)格式的申报资料,需包含临床试验数据库资料,并使用电子签章。
4、形式审查要点
1.申报事项审查要点:
● 若药物临床试验终止后需继续进行,需重新申请。
● 符合豁免条件的仿制药可直接申请上市许可。
● 临床价值明确的品种需依据《仿制药参比制剂目录》进行研发。
2.沟通交流审查要点:
● 申请附条件批准的药品需与药品审评中心沟通。
● 优先审评的申请需在提交上市许可申请前与审评中心确认。
3.申请表审查要点:
● 申请表信息应与证明性文件一致,特别是药品通用名称和商品名称。
5、申报资料审查要点
产品相关证明性文件:
提供合法来源证明文件包括供货协议、发票等适用于制剂未选用已登记原辅包情形)。
提供授权使用书(适用于制剂选用已登记原辅包情形)。如为供应商出具,需有原料药、药用辅料和药包材企业授权,并附授权书。
仿制药申请人需对照已在中国上市药品的专利信息进行声明。
临床试验相关证明文件:
包括药物临床试验批件和临床试验用药质量标准等材料。
6、其他提示
● 麻醉药品、精神药品等特殊药品不得委托生产。
● 境外生产药品的相关证明文件需符合世界卫生组织推荐的统一格式。
7、受理审查决定
1.受理:
符合形式审查要求的,出具《受理通知书》。
2.补正:
申报资料不齐全的,需一次性告知申请人补正内容。
3.不予受理:
不符合要求的,出具《不予受理通知书》。
8、附件
包括化学药品注册申报资料自查表和参考目录等。

来源:注册圈
关键词: 化学药品注册