
嘉峪检测网 2025-04-21 08:31
导读:本文介绍了如何快速解决液相色谱峰拖尾。
1. 前言
相信每一个做过液相色谱实验的小伙伴,都或多或少被“峰形拖尾”折磨过,峰拖尾是HPLC实验中最普遍的峰形问题,峰形的对称性直接影响定量分析的准确性。那么今天我们就来学习一下液相色谱峰拖尾的原因及解决方法。
(图片来源:参考资料【1】)
2 什么是峰拖尾?
首先,我们得要搞明白,什么是峰拖尾?
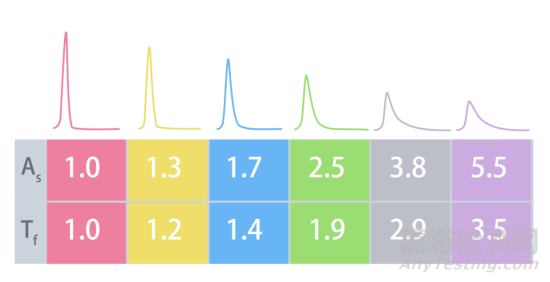
简单来说,就是色谱峰“头尖屁股宽”,峰的后半部分不对称了,也就我们所说的拖尾因子Tf>1.2。理想的情况是左右对称的高斯峰(Tf≈1),而拖尾严重时(Tf>2),就有可能造成两个拖尾峰连在一起,分不开,同时也会导致积分面积算不准,实验结果偏差大。

(图片来源:参考资料【1】)
3. 常见原因及解决方法
1.色谱柱内填料污染或塌陷
原因:长期使用色谱柱时,样品中的强保留物质(如蛋白质、脂类)会逐渐吸附在填料表面,就像“杂质糊住了筛网”一样,影响物质正常传质。此外,若操作中频繁使用极端pH(如pH<2或>8)或高温条件(>60℃),可能导致硅胶骨架溶解或键合相(如C18链)脱落,直接破坏填料结构,引发色谱柱填料塌陷。
解决方法:
冲洗再生:反向冲洗色谱柱(需确认柱子耐受反向压力)。
使用梯度溶剂冲洗:甲醇→乙腈→0.1M磷酸(pH 2)→纯水→高比例有机相(如95%乙腈)。
添加保护柱:在分析柱前加上一段保护柱,可以帮助拦截污染物,延长分析柱寿命。
更换新色谱柱:若柱效下降超40%或塔板数明显降低时,建议直接换柱。
2.色谱柱堵塞
原因:
若样品未经滤膜过滤(如未使用0.22 μm滤膜),其中的微小颗粒物(如细胞碎片、不溶杂质)就会直接堆积在柱头筛板上,阻碍流动相正常通过。此外,若流动相中的缓冲盐(如磷酸盐)未完全溶解或久置后析出结晶,也会卡在筛板孔隙中,进一步加剧堵塞。
解决方法:
反向冲洗疏通:用纯甲醇或乙腈反向冲洗(低流速,如0.2 mL/min)。
更换筛板:拆下柱头筛板超声清洗(异丙醇中超声10分钟)或更换新筛板。
预防措施:
○样品过0.22 μm滤膜,流动相过滤(0.45 μm)。
○缓冲液现配现用,避免盐结晶。
3.进样量过高
原因:
当样品进样量超过色谱柱的承载能力时(尤其是小内径柱或低载量柱),样品就会局部堆积,导致传质不均。这种过载现象会使色谱峰前沿陡峭、后沿拖拉,形成拖尾。
解决方法:
减少进样体积:常规分析柱(4.6×150 mm)建议进样量≤10 μL。
稀释样品:将样品浓度降低至线性范围内(如稀释10倍)。
换高载量柱:选择表面修饰更多或粒径更大的填料柱(如5 μm粒径替代3 μm)。
4.柱外效应(系统死体积过大)
原因:
如果色谱系统的管路过长、接头尺寸不匹配(比如粗管接细头)、流通池或进样阀体积过大,就会导致在色谱柱外的区域形成额外死体积。死体积过大会导致流动相流速变慢,样品在死体积处发生了滞留和扩散,从而形成峰形拖尾。
解决方法:
优化连接管路:
○使用内径≤0.13 mm的短管线(如15 cm以内)。
○更换匹配的PEEK接头(避免漏液和死体积)。
选择小体积检测池:流通池体积≤5 μL(如Agilent 1290 Infinity II的1.0 μL池)。
减少进样阀体积:使用低残留进样针(如10 μL定量环替代20 μL)。

(图片来源:参考资料【1】)
5.硅羟基效应(针对碱性化合物)
原因:
色谱柱硅胶表面残留的酸性硅羟基(-SiOH),就像“磁铁”一样,会牢牢吸住带正电的碱性化合物。二者通过离子交换或氢键作用,导致化合物滞留在柱子上,最终峰形拖尾。
解决方法:
添加扫尾剂:
○碱性物质:流动相加0.1%三乙胺(TEA)或二甲基辛胺(DMOA)。
○酸性物质:加0.1%三氟乙酸(TFA)或甲酸。
调节流动相pH:
○碱性化合物:pH调至高于其pKa 2个单位(如pKa=8,则pH=10)。
换用特殊色谱模式:
○亲水相互作用色谱(HILIC)或离子对色谱(IPC)。
4. 参考资料
【1】http://www.chromclass.com/article/technical-article/chromclass-discussion020/
【2】https://lab.jgvogel.cn/c/2023-11-10/1343090.shtml

来源:实验老司机
关键词: 液相色谱峰