
嘉峪检测网 2025-05-23 08:42
导读:仿制药开发需找到有区分力的溶出方法,通过有区分力的溶出方法不断提高自制制剂与参比制剂理化性质的一致性,再通过有限次数的预BE和BE数据才可能终结溶出、预BE、BE之间的反复拉扯。
引言
通过简单的溶出数据来预测药物制剂在复杂的人体胃肠道系统中的释放和吸收状态,这需要丰富的临床数据才能实现。由于高昂的临床成本,做仿制药开发很难获得丰富的临床数据。
为弥补这一遗憾,我们可以找到有区分力的溶出方法,通过有区分力的溶出方法不断提高自制制剂与参比制剂理化性质的一致性,再通过有限次数的预BE和BE数据才可能终结溶出、预BE、BE之间的反复拉扯。
1、溶出那些事
(1)相似因子f2与溶出相似度的关系
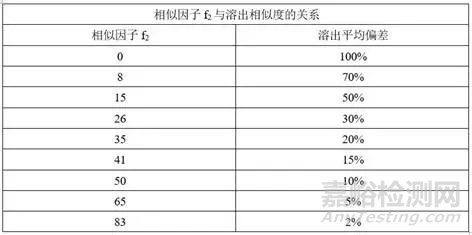
f2值越接近100,则认为两条溶出曲线越相似,一般情况下,f2值高于50,可以认为两条溶出曲线具有相似性。f2值达到多少可以放心的去做BE至今没有定论,药物研发人员对于f2值追求的脚步从未停歇。
f2值的大小需要结合产品的处方、工艺、体内释放、吸收进行系统性考察,一般情况而言:
速释制剂品种:重点考察低pH值介质(内控:相似因子f2值≥65),其次考察高pH值介质(内控:相似因子f2值>50);
缓释制剂品种:重点考察高pH值介质(内控:相似因子f2值≥65),其次考察低pH值介质的(内控:相似因子f2值>50);
最重要的是找到有区分力的溶出方法,重点做好最具区分力的那条溶出曲线,通过有区分力的溶出数据,调整自制制剂的处方、工艺,不断提高自制制剂与参比制剂理化性质的一致性,从而有效的提高自制制剂与参比制剂的溶出相似度。
(2)科学优化溶出方法
需要审慎的看待USP/FDA推荐的溶出方法,原研公司出于维护自身利益的角度出发,为了提高药物技术壁垒,公之于众的溶出方法大概率是经过调整的,可能阉割了溶出方法的区分力。
溶出方法的隐晦性在于无法证伪性,即使用USP/FDA推荐的溶出方法检测参比制剂,溶出结果必然是符合规定的,但是这个溶出方法的范围可能远大于BE通过的溶出范围,如果照此溶出方法开发自制制剂,可能导致自制制剂与参比制剂体外溶出完美拟合,但是最终自制制剂与参比制剂不具备体内等效性,所以我们需要审慎的使用公开的溶出方法。
需要重点警惕那些具有高转速、高浓度表面活性剂、高浓度有机溶剂及溶解度非常大的介质的溶出方法,如果使用这样的溶出方法开发制剂,制剂可能不到15分钟就迅速溶出85%以上,这样的溶出方法可能就是所谓的‘‘溶出参数烟雾弹’’了,制剂研究员需要科学的优化溶出方法。
优化溶出方法:
①降低转速(如:浆法:75s降至50s,篮法:100s降至75s、50s);
②减少溶出介质体积(如:900ml减少至500ml);
③非必要不使用表面活性剂,当需要使用表面活性剂时表面活性剂由低浓度缓慢增至高浓度(0.01%、0.02%、0.05%...);
④非必要不使用有机溶剂,当需要使用有机溶剂时有机溶剂由低浓度缓慢增至高浓度;
⑤优化溶出过程的取样点,合理分布(等量取样);
⑥警惕使用高溶解度的溶出介质,需要跟常规的溶出介质对比,不能超过漏槽条件过多(漏槽条件:是指药物所处释放介质的浓度远小于其饱和浓度,生理学解释为药物在体内被迅速吸收,为满足药物溶解-吸收的过程,漏槽条件起到了修正作用,一般释放介质的体积为药物饱和溶液所需介质体积的3~5倍);
⑦采用不同的溶出介质或试验条件进行溶出试验,包括但不限于USP/FDA推荐的溶出方法;
⑧除了速释制剂外(15分钟的溶出度大于85%),尽量使参比制剂和自制制剂在45min至120min之间溶出度达到85%,溶出速度不宜过快也不宜过慢;
(3)观察溶出现象
研究者应仔细观察溶出现象,分析溶出现象与溶出数据的关系,需要观察的现象包括但不限于以下内容:
①观察包衣片的包衣或胶囊的胶囊壳破裂的状态;
②观察制剂是否崩解完全并记录崩解时间;
③观察崩解出的颗粒是否有堆积现象;
④观察制剂是否存在黏连、粘壁,胶囊壳包裹颗粒等情况;
⑤观察自制制剂和参比制剂崩解出颗粒的大小和均匀性情况;
自制制剂的溶出现象应与参比制剂无显著差别,如果溶出现象差别较大,则需要结合溶出数据分析与制剂处方、工艺的关系。
(4)优化溶出方法
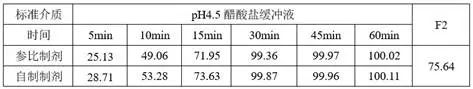
①优化前的溶出
1)溶出方法

2)溶出数据
②优化溶出方法
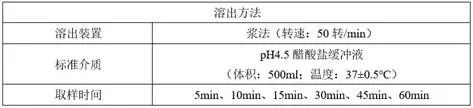
1)降低转速:浆法转速由75转/min降低至50转/min;
2)减少溶出介质体积:pH4.5醋酸盐缓冲液体积由900ml减少至500ml;
③优化后的溶出
1)溶出方法
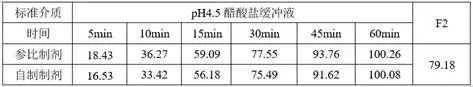
2)溶出数据
2、BE那些事
(1)评价生物等效性的适用范围
①平均生物等效性(ABE)
采用平均生物等效性的方法,生物等效性标准:受试制剂与参比制剂的主要药动学参数(AUC和Cmax)几何均值比的90%置信区间在80.00%~125.00%范围内。
②参比制剂标度的平均生物等效性(RSABE)
对于一些安全性较好、治疗窗较宽的高变异药物,在保证公众用药安全性、有效性的前提下和充分科学论证的基础上,通过部分重复或完全重复交叉设计,根据参比制剂的个体内变异,采用参比制剂标度的平均生物等效性的方法,将等效性判定标准在80.00%~125.00%的基础上适当放宽,可以减少不必要的人群暴露,从而达到科学评价不同制剂是否生物等效的目的。
(2)影响口服药物生物利用度的因素
口服药物的生物利用度取决于多种因素的共同作用,影响口服药物生物利用度的因素包括药物制剂的理化性质、人体胃肠道系统的生理状态。
①药物制剂的理化性质
1)药物制剂中活性成分的粒径、粒度分布、晶型;
2)药物制剂中功能性辅料:崩解剂、粘合剂、表面活性剂的种类、型号及用量;
3)药物制剂中功能性辅料对药物活性成分在体内外释放和吸收的影响;
4)药物制剂的关键工艺参数:崩解剂的加入方法、黏合剂的加入速度及加入量、制粒转速、制粒时间、颗粒的状态(休止角、松密度、振实密度、粒度分布)、片剂的硬度、包衣的厚度;
②胃肠道系统的生理状态
1)药物制剂中的活性成分在体内释放出来的速度和程度;
2)胃肠道中药物活性成分的吸收部位;
3)胃肠道中的吸收部位对药物活性成分的吸收能力;
4)药物制剂中释放出来的活性成分在吸收部位的停留时间;
5)胃排空和肠胃蠕动对药物活性成分吸收的影响;
6)空腹对药物活性成分吸收的影响;
7)餐后对药物活性成分吸收的影响;
自制制剂的药物活性成分在体内释放出来的速度和程度、胃肠道中的吸收部位对药物活性成分吸收的速度和程度应与参比制剂无显著差别,这需要通过保证自制制剂的理化性质与参比制剂的一致性来实现。
3、总结
首先采用有区分力的溶出方法不断提高自制制剂与参比制剂理化性质的一致性,其次通过对药物制剂中的活性成分在胃肠道系统中的释放和吸收的理解,再通过有限次数的预BE和BE数据才可能终结溶出、预BE、BE之间的反复拉扯。
药物研发人员对于如何提高BE通过率追求的脚步从未停歇。
4、参考文献
(1)国家食品药品监督管理总局《普通口服固体制剂溶出度试验技术指导原则》(2015 年 2 月).
(2)国家食品药品监督管理总局《普通口服固体制剂溶出曲线测定与比较指导原则》(2016 年 3 月).
(3)国家食品药品监督管理总局《已上市化学药品药学变更研究技术指导原则(试行)》溶出曲线研究的问答(2022 年11 月).
(4)国家食品药品监督管理总局《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》(2016 年 3 月).
(5)国家食品药品监督管理总局《人体生物等效性试验豁免指导原则》(2016 年 5 月).

来源:药事纵横
关键词: 药物研发