
嘉峪检测网 2025-06-19 16:52
导读:本文讨论了窄治疗窗药物的生物等效性要求,详见下文。
本文讨论了一种特殊药物,窄治疗窗药物的生物等效性要求。
一、窄治疗指数药物定义
根据2020年12月31发布的《窄治疗指数药物生物等效性研究技术指导原则》的定义。窄治疗指数(Narrow therapeutic index,NTI)药物或窄治疗窗药物一般是指剂量或血药浓度的微小变化即可能导致治疗失败和/或严重不良反应,进而危及生命,或者导致永久或严重的残疾或功能丧失的药物。
窄治疗指数药物通常具有以下特点:有效剂量与中毒剂量(或有效浓度与中毒浓度)接近;血药浓度低于有效浓度可能导致治疗失败,高于有效浓度可能导致严重不良反应;需要基于药动学或药效学指标进行治疗药物监测;具有较低或中等程度的个体内变异;临床应用中,剂量调整幅度通常较小等。
2019年11月8日的征求意见稿《已上市化学药品药学变更研究技术指导原则》附录三,公布了窄治疗指数药物目录,氨茶碱、茶碱、胆茶碱、双羟丙茶碱、苯妥因钠、丙戊酸、炔雌醇/孕酮制剂、地高辛、洋地黄毒甙、华法令钠、甲磺酸异他林吸入气雾剂、卡马西平、可乐定透皮贴剂、磷酸丙吡胺、硫酸胍乙啶、硫酸奎尼丁、硫酸哌唑嗪、硫酸异丙肾上腺素、米诺地尔、扑米酮、碳酸锂、盐酸克林霉素、盐酸可乐定、盐酸普鲁卡因胺、divalproex钠、左甲状腺素钠、环孢霉素A、他克莫司、西罗莫司、丙戊酸/丙戊酸钠。以上药物如无特别标明,一般指口服给药制剂。
二、窄治疗指数药物BE设计
与高变异药物不同的是,窄治疗指数药物BE研究总体设计只能是完全重复(两制剂、两序列、四周期)交叉设计。而高变异药物研究总体设计可以是完全重复交叉设计,也可以是部分重复(两制剂、三序列、三周期)交叉设计,特殊情况下(如长半衰期)还可以采用平行组设计。这是因为只有完全重复设计才能求出窄治疗指数药物BE判定的第三条指标,即比较受试制剂与参比制剂的个体内标准差。完全重复交叉设计,同一个受试者既吃两次参比制剂,又吃两次受试制剂,如下图。而部分重复交叉设计的受试制剂没有被吃两次,所以无法计算受试制剂的个体内标准差。
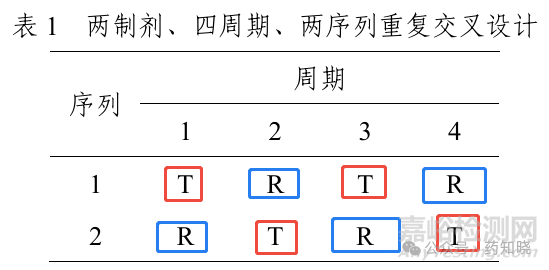
三、窄治疗指数药物BE判定标准
与一般化学药物相比,窄治疗指数药物进行生物等效性评价时,应采用更严格的等效性判定标准,以保证有效性和安全性。窄治疗指数药物等效性判定标准需要同时满足以下三个标准。
3.1、采用参比制剂标度的平均生物等效性方法(RSABE)评价等效性,即(YT-YR)2-θSWR2的单侧95%置信区间上限≤零。这一条判定标准与高变异药物相同,是ABE公式的变换形式,只是θ值有变化,其中△=1/0.9≈1.11,σw0=0.1,即θ≈1.11。
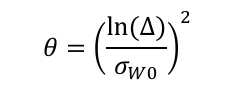
因为判定公式(YT-YR)2-θSWR2是平方形式,为正值,去掉平方后,会出现正负值。当受试比参比大时,为正值,当受试比参比小时,为负值。所以判定公式评价的仍然是经过参比制剂个体内变异校正的置信区间范围,即在90.00%-111.11%(变异10%时)的基础上调整,如下表和下图所示,变异越小的药物得到的置信区间范围越窄,分界线在CV%约为21.5%的地方。
| 分类 | 几何均值比 | 取对数 | 等效区间范围 |
|---|---|---|---|
| ABE | a/b | Ln(a/b) = Y<sub>T</sub>-Y<sub>R</sub> | (Y<sub>T</sub>-Y<sub>R</sub>)<sup>2</sup> < (Ln1.25)<sup>2</sup>,即:80.00% ≤ a/b ≤ 125.00% |
| RSABE | a/b | Ln(a/b) = Y<sub>T</sub>-Y<sub>R</sub> | (Y<sub>T</sub>-Y<sub>R</sub>)<sup>2</sup> < (Ln1.11)<sup>2</sup>(S<sub>WR</sub>/σ<sub>wo</sub>)<sup>2</sup>,即:90.00%·S<sub>WR</sub>/σ<sub>wo</sub> ≤ a/b ≤ 111.11%·S<sub>WR</sub>/σ<sub>wo</sub> |
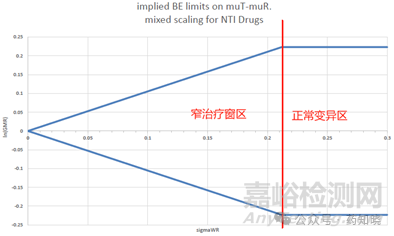
3.2、采用平均生物等效性方法评价等效性。这一条判定标准与高变异药物不完全相同,高变异药物要求的是点估计,而窄治疗窗药物要求的是区间估计。窄治疗指数药物这一条与常规药物评价标准一致,即受试制剂与参比制剂的主要药动学参数几何均值比的双侧90%置信区间应在80.00%~125.00%范围内。为什么增加这一条限定?主要目的是不希望拥有较大σWR的产品(≥0.213)置信区间超出80.00%~125.00%的范围。如下表所示,当变异系数CV%≥21.5%时,窄治疗指数药物采用参比制剂标度的置信区间范围将超限,若仅有等效性判定第一条,将无法保证窄治疗窗药物(变异大于21.5%产品)的安全性和有效性。
|
等效区间范围
|
SWRSWR | CV% | 参比标度的置信区间 |
|
(YT−TR)2<(ln1.11)2×(SWR/σwo)2(YT−TR)2<(ln1.11)2×(SWR/σwo)2 即:90.00% SWR/σwo<a/b<111.11% SWR/σwoSWR/σwo<a/b<111.11% SWR/σwo |
0.294 | 30 | 73.40%-136.24% |
| 0.213 | 21.5 | 79.93%-125.10% | |
| 0.198 | 20 | 81.17%-123.20% | |
| 0.149 | 15 | 85.46%-117.02% | |
| 0.1 | 10 | 90.00%-111.11% | |
| 0.05 | 5 | 94.87%-105.41% |
为什么针对这一条,高变异药物可以使用点估计,而窄治疗窗药物却采用区间估计?这是因为高变异药物第二条标准如果采用区间估计,会让第一条标准失去意义。如果高变异药物第二条标准的区间估计在80.00%~125.00%范围内,则和普通药物完全一致,第一条也一定能通过,也就失去了参比制剂标度的意义。
3.3、比较受试制剂与参比制剂的个体内标准差。相比高变异药物,这一条为新增标准。高变异药物仅要求与文献报道值的变异进行比较,并未明确差异的可接受度。而窄治疗窗药物除了要求与文献报道值进行比较外,还对受试制剂和参比制剂个体内标准差的比值进行了限定,即σWT/σWR的双侧90%置信区间上限应≤2.5。
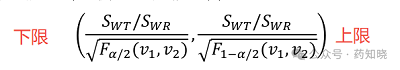
因为分母为F分布值,查表可得。样本量越大, 越小,因为
越小,因为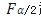 和
和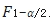 互为倒数关系,即
互为倒数关系,即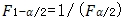 ,所以
,所以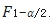
越大,即置信区间上限越小。这就是为什么增大样本量,也更容易等效的原因。
虽然高变异药物有两条判定标准,但是仅有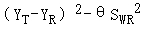 的单侧95%置信区间上限计算会用到参比制剂和受试制剂的标准差,能满足上限≤零的前提下,哪怕受试制剂的的标准差大一些,并不会引起安全性和有效性问题,所以可以不要求比较受试制剂和参比制剂的个体内标准差。
的单侧95%置信区间上限计算会用到参比制剂和受试制剂的标准差,能满足上限≤零的前提下,哪怕受试制剂的的标准差大一些,并不会引起安全性和有效性问题,所以可以不要求比较受试制剂和参比制剂的个体内标准差。
而窄治疗指数药物的前两条判定标准计算都会用到参比制剂和受试制剂的标准差。在两条标准都满足的前提下,如果受试制剂的变异明显比参比制剂大,受试制剂也有可能会引起安全性和有效性风险。这是因为窄治疗窗药物通常具有较低或中等程度的个体内变异,且对安全性和有效性的要求更高。
以样本量24为例,针对不同变异情况的药物,估算受试制剂与参比制剂个体内标准差理论最大差异,见下表。
|
CV% |
SWR |
SWT |
SWT-SWR |
置信区间上限(应<2.5) |
|---|---|---|---|---|
|
5 |
0.050 |
0.087 |
0.037 |
2.513 |
|
10 |
0.100 |
0.174 |
0.074 |
2.512 |
|
15 |
0.149 |
0.259 |
0.110 |
2.507 |
|
20 |
0.198 |
0.344 |
0.149 |
2.508 |
|
21.5 |
0.213 |
0.340 |
0.156 |
2.506 |
|
30 |
0.294 |
0.470 |
0.215 |
2.503 |
由上表可知,随着变异系数的增加,能够接受的受试制剂和参比制剂差异也越大。这一结论与高变异药物一致。
四、窄治疗指数药物BE计算步骤
4.1 计算参比制剂的个体内标准差
与高变异药物一样,采用以下公式计算,需要采用自然对数转化后的药动学参数计算。受试制剂的个体内标准差SWT计算方法相同。
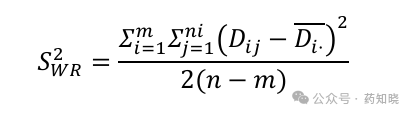
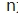 为受试者总人数(例如32例受试者),ni为第i个序列中受试者人数(总人数除以2,第1序列16人,第2序列16人),m为序列数(窄治疗窗为2序列),i为研究中的序列编号(第1序列和第2序列),j为序列内受试者编号(如编号001、002……032),Dij=Rij1-Rij2表示参比制剂前后吃两次的差值(对数转换后),
为受试者总人数(例如32例受试者),ni为第i个序列中受试者人数(总人数除以2,第1序列16人,第2序列16人),m为序列数(窄治疗窗为2序列),i为研究中的序列编号(第1序列和第2序列),j为序列内受试者编号(如编号001、002……032),Dij=Rij1-Rij2表示参比制剂前后吃两次的差值(对数转换后),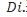 表示求该序列内前后吃两次差值的平均值(1和2序列应分别计算)。
表示求该序列内前后吃两次差值的平均值(1和2序列应分别计算)。
4.2 计算以下算式的单侧95%置信区间上限:
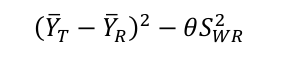
(YT-YR)2-θSWR2的单侧95%置信区间上限应≤零。其中YT和YR分别表示受试制剂和参比制剂自然对数转换后的AUC或Cmax的均值。θ已知约为1.11,SWR在上步已经求出。在上篇文章《如何让高变异药物更容易BE等效》中介绍了计算方法,置信区间上限U的计算参照下面公式,U=点估计值+估计误差。需要知道三个数据:受试制剂和参比制剂Cmax/AUC取自然对数后的平均差(YT-YR)、参比制剂和受试制剂的个体内标准差SWR和SWT、参比制剂和受试制剂的样本量nR和nT。
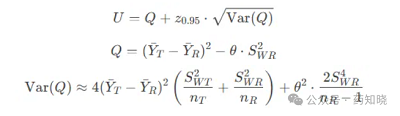
按照3.3项下,将受试制剂和参比制剂的个体内差异取到上限附近,假设样本量24,满足等效情况下,受试样品和参比制剂药动参数几何均值比的最大取值范围见下表。
|
CV% |
SWR |
SWT |
SWT/SWR 置信区间上限(应<2.5) |
几何均值置信区间 |
|---|---|---|---|---|
|
5 |
0.050 |
0.086 |
2.485 |
98%-102% |
|
10 |
0.100 |
0.173 |
2.497 |
95%-105% |
|
15 |
0.149 |
0.258 |
2.497 |
93%-107% |
|
20 |
0.198 |
0.343 |
2.500 |
91%-110% |
|
21.5 |
0.213 |
0.368 |
2.499 |
90%-111% |
|
30 |
0.294 |
0.508 |
2.498 |
87%-115% |
由上表可知:如果受试制剂和参比制剂两者的个体内变异相差较大(一般是制剂质量因素导致),与正常变异情况的3.2项表对比,受试制剂和参比制剂药动参数的比值范围将缩小,这就增加了BE等效难度。
4.3 计算平均生物等效置信区间:
与常规药物的计算方法一致,采用ABE方法计算受试制剂和参比制剂的主要药动学参数几何均值比的双侧90%置信区间,应在80.00%~125.00%范围内。需要注意的是,对数转换后,仍然不能按照求平均值的方式用excel直接计算,需要采用混合效应模型的最小二乘法计算,通常需要专业的统计软件,如Phoenix WinNonlin或SAS。
最小二乘法,我们做分析方法学的线性时会用到,只不过分析方法只有一个变量,就是浓度因素,属于一元线性回归;而BE试验有多个变量,如制剂、周期、序列、受试者等因素,属于多元线性回归。
4.4 计算受试制剂与参比制剂个体内标准差比值置信区间:
受试制剂与参比制剂个体内标准差比值(σWT/σWR)的双侧90%置信区间(即α=0.1)计算如下式:
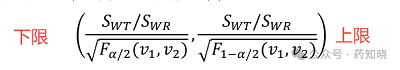
SWT和SWR在4.1步中均已经求出来,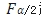 值可以通过查表得到,
值可以通过查表得到,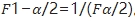 。不同例数的F理论值见下表,最后一列作为分母用来求置信区间上限,因为分母小于1(在0.5附近),所以受试制剂的变异通常不会比参比制剂的变异大1.5倍。
。不同例数的F理论值见下表,最后一列作为分母用来求置信区间上限,因为分母小于1(在0.5附近),所以受试制剂的变异通常不会比参比制剂的变异大1.5倍。
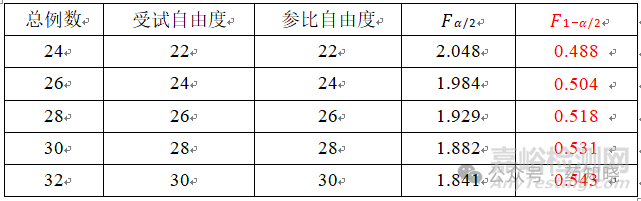
自由度一般是总例数减去2,需要注意的是,在正式试验中,因受试者脱落的影响,受试组和参比组的自由度可能会不相同。
4.5 判定等效性:
只有主要药动学参数(AUC0-t、AUC0-∞和Cmax)同时符合上述三个等效性判定标准(即采用RSABE方法评价等效性、采用ABE 方法评价等效性和比较受试制剂与参比制剂的个体内标准差),才可判定受试制剂与参比制剂具有生物等效性。
五、结语
对于药学开发人员来说,对BE试验和数据接触的相对较少,为了便于理解,我们拿溶出曲线做个类比。当溶出曲线各时间点数据RSD符合要求时,采用f2比较相似性,f2比较采用的是每个时间点的均值,就是点估计。当RSD不符合要求时,推荐采用bootstrap法比较相似性,bootstrap法比较,通过模拟重复抽样,得到每个时间点围绕均值的上下波动限度,也就是置信区间。f2法和bootstrap法基于相同的统计逻辑,而多变量置信区间法(MSD)逻辑则不同。受参比制剂批次限制,采用MSD方法,很多时候参比制剂自身也很难相似,这也是为什么ICH《M13B:其他规格的生物等效性豁免》中已经不再提到MSD法的原因。有兴趣了解细节的朋友,可以阅读往期文章《溶出曲线RSD偏大之“多变量置信区间法”》。
一般药物BE试验的ABE判定标准,我们可以理解为点估计和区间估计都要在80.00%~125.00%范围内。高变异药物BE试验的RSABE判定标准,可以理解为点估计在80.00%~125.00%范围内,区间估计在参比制剂校正后的范围内。窄治疗指数药物BE试验的RSABE判定标准,可以理解为区间估计既要在ABE的范围内,又要在参比制剂校正后的范围内,且两制剂的变异情况不能相差太大。通过高变异药物和窄治疗窗药物BE逻辑分析,我们发现BE难度随着变异系数减小而上升,这是因为变异系数越小,要求受试制剂和参比制剂之间的差异也越小。这如同压片一样,片重越小的药片,对物料属性和压片机精度要求越高,越难控制。理解BE试验数据统计的底层逻辑,有助于药品开发人员提升质量源于设计(QbD)理念的理解,提高自己对药品质量的把控度,真的做到仿制药与参比制剂质量和疗效一致性。

来源:药知晓