
嘉峪检测网 2025-06-30 08:43
导读:结合近年上市的几个具体案例看下IV改SC的非临床开发思路。
很多抗肿瘤生物药初始上市给药途径是静脉注射(IV),后续为提高给药便利性,会开发皮下(SC)给药途径,那么从IV切换到SC会涉及哪些非临床研究呢?不同群友,观点不一。有说要重新开展全面的新给药途径药理毒理研究?有说可以考虑开展简化的毒理试验,比如单一种属或组别或恢复期优化?也有说如果IV给药非临床暴露量可以覆盖SC,是不是开展局部毒性即可?关于这个问题从三个角度尝试做下讨论,一是FDA于2015年颁布的《Nonclinical Safety Evaluation of Reformulated Drug Products and Products Intended for Administration by an Alternate Route》,里面对于制剂变更和给药途径变更给出了基本原则;二是NMPA早年发表过一些电子刊物,如《改良型新药非临床研究的一般考虑及需要关注的问题》、《桥接研究在药物非临床研究与评价中的应用》等;三是目前已经有多款已上市IV给药生物制品增加SC给药剂型,可作为案例参考。
先看下FDA指导原则给出的建议。三个关键词—暴露量、给药周期、给药途径。如果单纯制剂处方变更,但给药周期、给药途径不变,前期非临床暴露量能覆盖新制剂,需要补充的非临床研究很少,视具体情况,可能局部毒性或者短期重复给药毒性试验即可支持上市(For a new formulation that will be used by the same route as the previously approved product, shorter duration toxicity studies than those outlined in ICH M3(R2) may be appropriate)。如果给药途径出现变化,无论制剂处方变更与否,无论暴露量是否覆盖,均需要补充开展非临床研究。那么具体需要开展哪些非临床研究呢?
首先,要看已批准制剂的非临床研究是否符合当前标准,如果不符合,新的非临床研究既要评估新给药途径的安全性,还要弥补之前的缺陷。比如有些药物上市时间早,上市后很多年才涉及给药途径或制剂变更,早年的毒理终点指标可能不完善。
其次,For all drug product reformulations and for all drug products with new routes of administration, acute and/or repeat-dose toxicity studies with complete histological evaluation should be conducted using the clinical route of administration. Acute studies may not be needed when repeat-dose studies are conducted. In general, durations of the toxicity studies should follow the recommendations outlined in ICH M3(R2) or ICH S9.更换新给药途径一般需要开展急性和/或重复给药毒性试验,伴随开展组织病理学检查。如果开展重复给药毒性试验,可以不做急毒。给药周期参考ICH M3(R2)或ICH S9规定。那么,新给药途径毒理研究有简化空间吗?还是按照传统标准研究,如果有两个相关动物种属,需要在两个种属中均开展毒理试验,需要设置4个组别(vehicle+3剂量),需要开展完整的毒理终点评估?
关于毒理终点,FDA指南中仅涉及组织病理学检查的描述,If systemic exposure by a new route of administration is equivalent to or less than that of the approved route, histological evaluation may be limited to locally exposed tissues.即如果新给药途径的暴露量≤原给药途径,仅开展局部组织的病理即可。NMPA 2008年孙涛等发表的文章《化学药口服改静脉注射给药非临床安全性研究评价要点》中提到,对于化学药物口服改静脉,如果新途径下Cmax/AUC不高于原途径,则开展进一步非临床评价的意义不大。不过,该文章已经发布16年,不确定是否代表当下监管思路,也不确定是否能借鉴到生物药静脉改皮下这个场景,毕竟皮下给药的Cmax/AUC通常低于静脉。
关于动物种属,FDA 2008年草案中其实有提及,根据给药途径的不同,或用单一种属动物(例如,经皮给药制剂或贴片的皮肤;应用吸入给药制剂的肺脏;应用口服给药制剂的胃肠道;应用静脉注射、皮下注射、腹腔内注射、或者肌内注射给药制剂的注射部位;缓释注射或者植入给药制剂;海绵体内或尿道内给药;膀胱内给药),或用两个种属动物(例如,眼部给药,椎管注射或硬膜外注射)。但是,在2015年最终的指导原则中,这段描述不见了,不确定是各方未达成一致,还是很多给药途径开展单一种属毒理研究不能覆盖所有情况。总之,这代表早年监管机构的一个思路。
NMPA孙涛、王庆利2014年发表的《桥接研究在药物非临床研究与评价中的应用》一文中提出,对于改变给药途径的药物,非临床研究的考察重点是药物改变给药途径后物质基础的变化,包括与原给药途径相比药物暴露形式是否发生改变,药物的吸收、暴露量、组织分布等是否有所改变,是否形成新的代谢产物,药动学的改变是否会带来有效性和/或安全性的改变。因此非临床桥接研究首先进行与原给药途径的药动学比较,根据比较的结果设计后续药效学和/或毒理学研究项目。对于生物利用度或暴露量降低的情况,应重点关注改变给药途径后有效性是否能满足治疗需要。反之应关注改变给药途径后药物的毒性是否增加。对于组织分布改变和形成新代谢产物的情况,亦应重点考察安全性方面的变化,关注是否出现新的毒性靶器官或新的毒性反应。
解读下,按照CDE这个逻辑,IV改SC,生物利用度或暴露量通常会降低,应该更关注有效性是否满足。按照FDA的逻辑,改变给药途径通常需要开展毒理研究,新途径暴露量低于原途径,可以简化试验设计。综合来看,IV和SC两种给药途径的药代动力学对比研究是有必要的,对比暴露量变化。药效对比和简化的毒理研究也需要开展。
结合近年上市的几个具体案例看下IV改SC的非临床开发思路。
案例一:Rituxan and Rituxan Hylecta
Rituxan是一款靶向CD20的人鼠嵌合单抗,静脉注射给药,于1997年11月26日被FDA首次获批上市,先后获批了成人、儿童非霍奇金淋巴瘤,成人慢性淋巴细胞白血病等适应症。给药方案根据适应症和联用药物不同有很大区别,剂量从250mg/m2到1000mg/人不等,给药频率有QW、Q2W、Q4W等多种方式。Rituxan Hylecta是其皮下制剂,2017年6月22日上市。Rituxan Hylecta是Rituxan与重组人透明质酸酶共制剂,后者可以降解皮下的透明质酸,促进前者的吸收。Rituxan Hylecta临床给药剂量1400mg-1600mg/人。从IV给药的Rituxan到SC给药的Rituxan Hylecta,开展了哪些非临床支持性研究呢?
二者虽然制剂处方不同,Rituxan Hylecta多了透明质酸酶这一活性成分。不过,单独的Rituxan(1997年)和透明质酸酶(2005年)均已经上市。
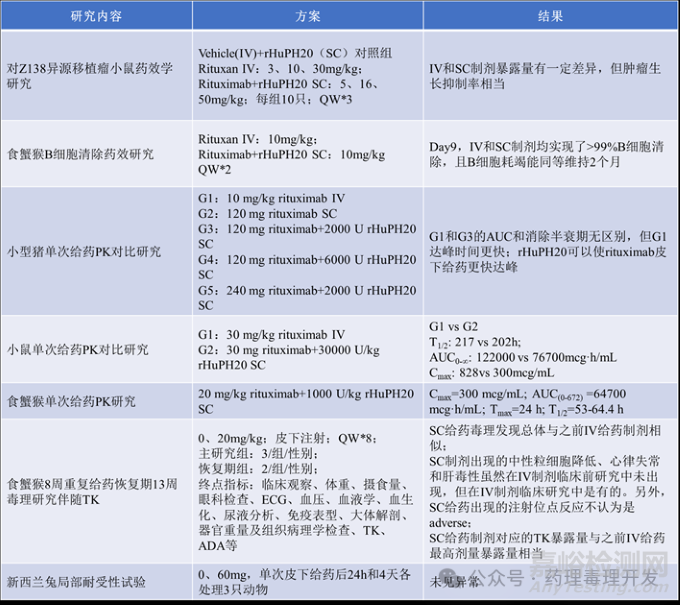
为支持Rituxan Hylecta上市,额外开展了非临床药理学、药代动力学和毒理学研究,同时桥接已上市Hylenex、Rituxan两个个产品的资料。Rituxan Hylecta应该是首个含透明质酸酶的共制剂产品,围绕透明质酸酶和Rituxan+透明质酸酶共制剂开展了大量药理毒理研究。篇幅所限,透明质酸酶相关研究暂且不表,重点看下围绕Rituxan Hylecta整体制剂开展了哪些非临床研究。主要包括2项主要药效学研究,3项药代动力学研究和2项毒理学研究。
药效学研究主要考察了IV制剂和SC制剂在抗肿瘤药效和PD指标方面的可比性。小鼠Z138 CDX模型证明了二者虽然暴露量略有差异,但SC制剂剂量提高60%即可实现药效与IV制剂的可比。食蟹猴PD研究则证明同样剂量下,两种制剂清除B细胞的能力相当。
药代动力学对比研究显示,SC制剂剂量提高40%可以实现与IV制剂AUC和消除半衰期的一致性(小型猪120mg约等于14mg/kg)。
毒理研究则证明高剂量SC给药毒理学表现与前期IV制剂结果一致。家兔SC给药局部耐受未见异常。
所以,SC制剂临床剂量较原IV制剂提高40%-60%,实现同样治疗效果,未见新的安全性方面的担忧。
案例二:Herceptin and Herceptin Hylecta
Herceptin是一款靶向HER2的单抗,静脉注射给药,于1998年9月25日被FDA首次获批上市,先后获批了HER2过表达乳腺癌辅助治疗、转移性HER2过表达乳腺癌和HER2过表达胃癌适应症。首剂量4mg/kg,之后2mg/kg,每周给药1次。或者首剂量8mg/kg,之后6mg/kg,每3周给药1次。Herceptin Hylecta是其皮下制剂,2019年2月28日上市。Herceptin Hylecta是Trastuzumab与重组人透明质酸酶共制剂,后者可以降解皮下的透明质酸,促进前者的吸收。Herceptin Hylecta每3周给药1次,每次2-5分钟,给药剂量600mg/人,如果用于乳腺癌辅助治疗,应至少给药52周或至疾病复发,以先出现为准。如果用于转移性乳腺癌治疗,给药直至疾病出现进展。可以看出Herceptin Hylecta在乳腺癌中的适应症与Herceptin一致,但未申请胃癌适应症,主要原因是PK差异。从IV给药的Herceptin到SC给药的Herceptin Hylecta,开展了哪些非临床支持性研究呢?
二者虽然制剂处方不同,Herceptin Hylecta多了透明质酸酶这一活性成分。不过,单独的Herceptin和透明质酸酶均已经上市,且CD20单抗Rituxan Hycela也使用过透明质酸酶作为制剂组分。言外之意,不需要针对制剂辅料开展额外的非临床研究。
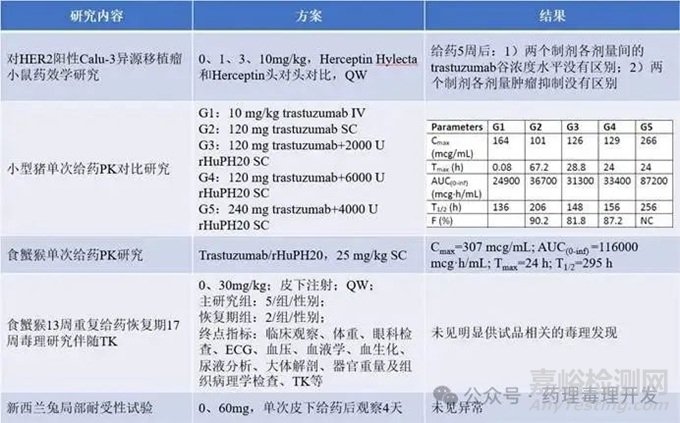
为支持Herceptin Hylecta上市,额外开展了非临床药理学、药代动力学和毒理学研究,同时桥接已上市Hylenex、Herceptin、Rituxan Hycela三个产品的资料。Herceptin Hylecta试验资料总结如下表所示,一共开展了5项研究,药理学试验1项,药代动力学试验2项,毒理学试验2项。
药效试验的目的是确认皮下制剂和静脉制剂同样剂量抗肿瘤效果是否类似,每种制剂开展了3个剂量的对比,结果证明二者在各剂量下的谷浓度相似且抗肿瘤效果一致。
小型猪药代动力学试验结果显示,加入透明质酸酶的皮下制剂吸收更快(平均Tmax=24-29h vs 67h),加入透明质酸酶对药物的暴露量如Cmax和AUC0-inf,或者生物利用度没有影响。
食蟹猴重复给药毒理研究结果显示,皮下给药制剂与之前静脉制剂毒理结果一致,均未见明显供试品相关的异常。值得一提的是,本研究较传统设计进行了简化,仅设计了1组给药组。
兔局部耐受性试验也未见异常。
案例三:Darzalex and Darzalex Faspro
Darzalex是一款靶向CD38的单抗,静脉注射给药,于2015年11月16日被FDA首次获批上市,用于多发性骨髓瘤的治疗。给药剂量16mg/kg。Darzalex Faspro是其皮下制剂,2020年5月1日上市。Darzalex Faspro是Darzalex与重组人透明质酸酶共制剂,同样用于多发性骨髓瘤治疗,给药剂量1800mg/人。
Darzalex Faspro开展的非临床研究比较有限。主要因为daratumumab仅与人和猩猩的CD38结合,没有合适的相关动物种属开展毒理研究。Darzalex Faspro仅开展了兔和小型猪皮下给药局部耐受试验。FDA同意并接受了企业的观点。Darzalex Faspro最终临床剂量是通过与静脉给药制剂非劣临床对比研究确定,证明1800mg/人的Darzalex Faspro与16mg/kg的静脉给药制剂Darzalex药效相当(ORR,41.1% vs 37.1%),谷浓度(Ctrough at pre-dose of Cycle 3 Day 1)也相当。
案例四:TECENTRIQ and TECENTRIQ HYBREZA
Tecentriq是一款靶向PD-L1的抗体,开发了静脉和皮下两种给药途径。静脉给药制剂2016年5月18日上市,皮下给药制剂2024年9月12日上市。
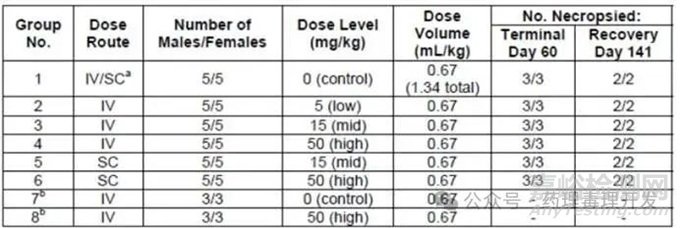
支持皮下制剂上市的药理毒理研究中,未开展药理学研究,而是参考的静脉给药制剂的上市资料。静脉制剂仅采用替代抗体开展了腹腔注射给药的抗肿瘤药效学研究。FDA接受不需要额外开展皮下制剂的药理学研究。同样的,皮下制剂也未额外开展非临床ADME/PK研究。当然,也未开展遗传毒性、生殖毒和致癌性试验。仅开展了一般毒理研究。不过,这项毒理研究并不是专为皮下制剂而设立。实际上是在开发静脉给药制剂时开展的食蟹猴重复给药毒理研究中,带上了两组皮下给药,如下表所示。结果证明,本品皮下和静脉给药毒性学研究结果相似。FDA接受这一毒理学设计和结果。
' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
最后
Rituxan Hylecta和Herceptin Hylecta的思路还是比较相似的,基本按照单模型药效对比+多种属PK对比+简化的一般毒理试验+局部耐受性的思路开展的研究,即暴露量未增加,药效等效,系统毒性未增加,给药局部可耐受。Tecentriq Hylecta则要简化很多,仅开展了一般毒理/TK试验,主要引用的Tecentriq和Hylenex的资料。

来源:药理毒理开发
关键词: 生物药物静脉给药