
嘉峪检测网 2025-08-18 10:03
导读:近年来,抗体-药物偶联物(ADC)的获批从根本上改变了某些血液系统恶性肿瘤和实体瘤的治疗模式与标准治疗方案。
近年来,抗体-药物偶联物(ADC)的获批从根本上改变了某些血液系统恶性肿瘤和实体瘤的治疗模式与标准治疗方案。ADC将单抗的特异性和细胞毒类药物的高活性通过连接子结合在一起,旨在提高细胞毒性药物的治疗指数。这类产品在设计非临床到临床的转化策略时,有几项重要考虑因素。首先,动物模型在预测人体对ADC反应方面的相关性。其次,ADC的PK与PD之间的相互作用——即药物在靶点处的浓度如何影响治疗结果,这方面需要复杂的建模技术。最后,理解ADC的免疫原性潜力,包括抗药抗体的生成,对于预测其临床疗效和安全性同样重要。
鉴于ADC的独特结构和作用机制,其非临床安全性评估较为复杂。ADC的安全性不仅取决于药物的药理学作用,还与其生物物理和生化特性、免疫原性风险以及非靶向作用有关。这种复杂性要求对ADC的各个组成部分(抗体、连接子和载荷)有深入的了解,因为它们对ADC的毒理学特点分别有不同的贡献。非临床PK/PD特征对于ADC同样重要。ADC的PK特征受抗体组分的稳定性、解离、连接子的可切割性以及载荷的物理化学性质的影响。因此,非临床PK/PD研究必须精心设计,以表征ADC的体内特性,然后利用定量建模来预测人体的PK行为、生物分布和ADC的消除。定量PK/PD建模整合了各种非临床研究的数据,可用于有效剂量预测和某些安全风险缓解。
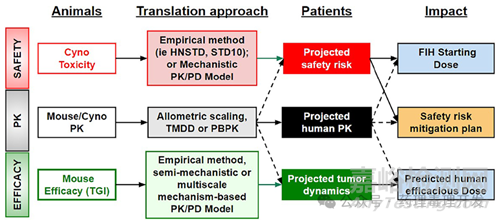
ADC开发是一项多学科的工作,需要将非临床安全性、药理学和PK/PD建模战略性地整合在一起,以成功地从实验室转化为临床应用。ADC开发所涉及的复杂性强调了对强大药理学和疾病理解的需求,借助先进的分析和建模工具来预测人类反应并优化治疗结果。本文旨在提供一个ADC 的PK/PD转化框架(如下图所示)。
分析方法/分析物
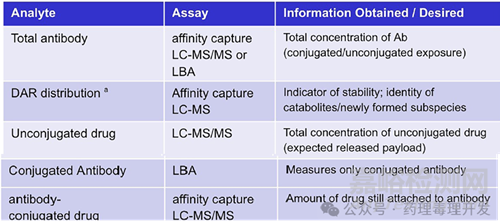
ADC药物具有复杂的分子结构,融合了小分子药物和大分子药物的特性。根据偶联方法的不同,ADC药物产品可以是均质或异质的混合物,这些混合物不仅在于连接的载荷分子数量不同,载荷连接的蛋白结合位点也可能存在差异。此外,ADC在体内的降解和代谢过程会进一步增加其复杂性。这会导致初始载荷/抗体比(DAR)分布的改变,原因是药物载荷的脱落,以及新药物形式的产生,如代谢物、连接子和连接子-药物、与内源性分子(如白蛋白、半胱氨酸)形成的加合物,以及与可溶性/脱落的靶抗原和其他抗体形成的复合物。由于DAR分布可能在体内持续变化,并可能影响ADC的清除、分布和活性,因此建立合适的检测方法至关重要。通常,利用亲和捕获LC-MS生物分析法来表征DAR分布。除了DAR分布外,这些分析方法还可以提供关于药物载荷脱落机制的证据,例如对于采用马来酰亚胺化学偶联的ADC,会发生马来酰亚胺交换以及与白蛋白或半胱氨酸形成加合物。
鉴于ADC给药后浓度和组成的异质性和复杂变化,通常在非临床和临床研究中测量3种不同的分析物,以表征ADC的药代动力学特性,即偶联物(以偶联载荷或偶联抗体测量)、总抗体(完全偶联、部分偶联和未偶联抗体)和未偶联载荷,如下表所示。
PK转化
对于单抗,典型的PK预测方法通常是基于单一种属进行异速缩放,其中以猴为主。单抗基于体重的缩放指数范围为0.75到1。相比之下,小分子药物的人体PK预测涉及多种放大方法,包括基于代谢的缩放、多种属异速缩放、含校正因子异速缩放以及基于指数规则的异速缩放。不过,考虑到ADC的PK主要由其抗体部分主导,故缩放策略主要参照单抗。
有研究预测了11种MMAE/DM1 ADC人体PK的预测方法。这些方法包括使用猴的PK数据以及几种多种属缩放方法,主要针对ADC的两个成分:偶联物和总抗体。大多数方法要么低估,要么高估了人体的清除率(CL),但使用猴的PK数据和CL的缩放指数为1进行的预测的最为准确,而且这一结果在偶联物和总抗体中是一致的。
不过,与微管蛋白抑制剂不同,DNA损伤剂,无论是直接与DNA结合(如Loncastuximab tesirine),还是通过拓扑异构酶I抑制间接作用(如Trastuzumab deruxtecan和Sacituzumab govitecan),其PK转化则不那么明确。对于Loncastuximab tesirine,其转化为人的PK接近1,与微管抑制剂类似。对于Trastuzumab deruxtecan和Sacituzumab govitecan,则比较有趣。这两种ADC均通过链间二硫键实现高偶联率(DAR,7-8个载荷/单抗),分别使用可切割的连接子mc-GGFG-AM和CL2A。对于Trastuzumab deruxtecan,基于从猴预测人体清除率比实际观察到的更慢。相反,Sacituzumab govitecan在人中的清除率与预测相似或略快。对于这些新型高DAR ADC(包括对连接子化学差异的理解),需要更多数据来了解从非临床到临床的PK转化。
首次用于人体(FIH)的剂量计算
鉴于ADCs结构复杂,包含大分子和小分子成分,因此在估算FIH剂量时,兼顾小分子药物和生物制品策略。Sabre等人在对20个ADC的IND评估时发现,FIH剂量确定方法虽然主要基于毒理学终点,但也呈现一定的多样性。这些标准包括采用典型的小分子毒理学终点,例如在10%动物引起严重毒性的剂量(STD10),以及采用大分子毒理学终点,例如最高非严重毒性剂量(HNSTD)或未观察到不良效应水平(NOAEL)(Saber和Leighton,2015)。
同样,在确定ADCs的人体等效剂量时,也采用了不同的换算方法。对于小分子药物,通常采用体表面积(BSA)进行换算;而对于大分子药物,则常采用体重(BW)进行换算。最终,Sabre等人发现,基于食蟹猴HNSTD的1/6或啮齿动物STD10的1/10,并根据体表面积(BSA)进行换算,通常能够在Ⅰ期临床试验中实现安全且有效的剂量递增。
不过,Sabre等人的这项研究中,ADCs的载荷主要集中在美登素或单甲基奥瑞他汀,对于其它载荷是否也适用呢?Saber等人对含其他载荷的ADC(如DNA损伤剂)的FIH起始剂量也进行了研究,与美登素/奥瑞他汀类不同,建议采用更大的安全系数(如1/10 HNSTD),以确保患者安全。对于其他一些较老的ADCs,如Gemtuzumab ozogamicin和Inotuzumab ozogamicin,其FIH剂量的确定采用了NOAEL(EMA,2008,2017)。此外,有时还会采用PK/PD模型来确定剂量。
临床有效剂量预测
近年来,针对ADCs预测人体有效剂量的方法不断发展,从基于体重的经验方法(Griffin等人,2022),到semi-mechanisticPK/PD建模方法(Li等人,2023),再到more mechanism-based的方法(如multiscale mechanistic PK/PD或定量系统药理学建模方法)(Scheuher等人,2023)。
Griffin等人提出使用种属间体形大小(如体表面积或体重)将小鼠有效剂量换算为临床有效剂量(Griffin等人,2022)。研究显示,基于体重的剂量换算方法能够较为合理地预测人体有效剂量。在9种ADC中,有6种(77.7%)的预测值在临床有效剂量的2倍和3倍范围内。与此一致,在12种获批的ADC中,使用基于体重的方法,分别有67%和83%的ADC的临床有效剂量预测值在临床批准剂量的2倍和3倍范围内。这种方法所需数据较少,仅需相关小鼠异种移植模型中的有效剂量数据,但其预测准确性通常低于其他两种方法。该方法通常用于药物发现的早期阶段,用于优先选择分子并评估剂量可行性,以避免进一步开发那些可能需要过高临床剂量的ADC。
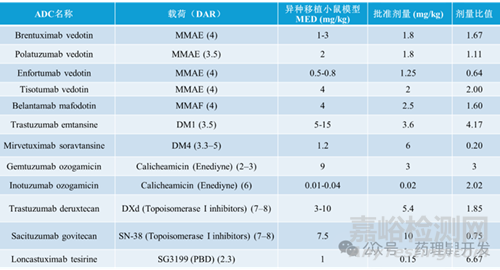
Jumbe等人开发了一个semi-mechanistic肿瘤生长抑制(TGI)模型,用于描述小鼠异种移植模型中对T-DM1的抗肿瘤反应(Jumbe等人,2010)。该模型包含多个肿瘤传递室,以考虑ADC药代动力学与肿瘤生长抑制之间的延迟,药物的杀伤效应由外周ADC浓度引发。为了将PK/PD模型从小鼠转化为人体,小鼠的PK参数被预测的人PK参数所替代,而估计的药效学参数则假设在小鼠和人类之间是相同的。人体PK参数可以通过从食蟹猴的PK数据进行异速缩放预测。通过模型模拟不同剂量和方案以实现疾病稳定(即肿瘤停滞),可以估计人类有效剂量和方案。由于癌症患者的肿瘤倍增时间远慢于小鼠异种移植模型,这种方法比较保守,使用PK/PD转化模型实现疾病稳定的预测被认为在人体中至少是最低限度有效的,可能实现肿瘤缩小(Betts等人,2020)。该模型已通过T-DM1有效剂量预测的临床验证(Haddish-Berhane等人,2013),并被用于将PF-06804103(Betts等人,2020)和一种针对恶性实体瘤的间皮素靶向ADC从临床前转化到临床(Li等人,2023)。
值得注意的是,上述PK/PD转化模型是适合特定目的且semi-mechanistic的。该模型只需要药代动力学和体内肿瘤生长/缩小数据,并在经验的基于体型大小的方法和full-fledge mechanistic模型(如multiscale定量系统药理学模型)之间提供了一种折衷。它在从小鼠模型转化为人体时,没有考虑种属间在靶点表达/分布、ADC及其载荷在作用靶部位的PK、肿瘤穿透、肿瘤生长速率和异质性方面的差异。Shah等人开发了一个multiscale mechanistic ADCs的PK/PD模型,该模型整合了多个维度,以实现ADC疗效从临床前到临床的转化(Shah等人,2012)。该模型包括四个子模块:1)一个药代动力学模型,描述ADC及其释放载荷在系统循环中的代谢和清除;2)一个肿瘤相关模型,描述ADC扩散到肿瘤情况;3)一个细胞模型,描述ADC与靶点的结合、内吞、细胞内载荷释放、载荷与靶点的结合、从肿瘤细胞外排或扩散(如果有旁观者效应);4)在小鼠异种移植模型中,由肿瘤载荷浓度引发药物杀伤效应的肿瘤生长抑制。通过纳入人体药代动力学以及相关患者人群中的临床相关肿瘤体积、肿瘤生长速率和靶点表达,将建立的临床前模型转化为临床应用。这种模型已成功预测了brentuximab vedotin(Shah等人,2012)、inotuzumab ozogamicin(Betts等人,2016)、T-DM1和trastuzumab deruxtecan(T-DXd)的临床疗效(Scheuher等人,2023)。尽管该模型较为复杂,开发需要更多时间和资源,但其建模框架被认为可以通过替换相关参数来适应其他ADC,并用于促进ADC设计、候选药物选择、临床有效剂量预测、FIH剂量递增选择以及临床剂量方案优化。
PK/PD安全性转化
血液学毒性(如血小板减少症和中性粒细胞减少症)是ADC药物常见不良事件,通常需要进行剂量调整(暂停或减少剂量),直到血小板或中性粒细胞恢复。为了表征ADC给药后血液细胞计数的动态变化,已开展了许多临床和临床前PK/PD模型的建立工作。
例如,Ait-Oudhia等(2017)开发了临床semi-mechanistic PK/PD模型,用于表征T-DM1和PCA062(抗P-钙粘蛋白-DM1 ADC)对患者血小板计数的影响,能实现准确描述血小板计数的动态变化以及3级及以上血小板减少症的发生率。同样,Ait-Oudhia等(2017)还开发了一个临床前PK/PD模型,用于研究小鼠中T-DM1的血小板减少症和中性粒细胞减少症。通过建模,评估了ADC及其相关成分(如总抗体、偶联抗体和未偶联载荷)的血药浓度和血液毒性的关系。结果显示,T-DM1的浓度是导致血小板减少症和中性粒细胞减少症的物质基础,与临床一致。
对于从临床前动物模型到人体的骨髓抑制外推,建议对小分子抗癌治疗药物进行种属差异校正,包括蛋白结合和通过粒细胞/巨噬细胞集落形成单位(CFU-GM)实验确定的敏感性。比如,MMAE血浆蛋白结合就具有种属依赖性,大鼠和人类的结合水平较高(67.9%-82.2%),而小鼠和猴的结合水平较低(17.1%-28.5%)。
Jie等利用4种不同的vc-MMAE ADC治疗后的PK和PD数据,探索了基于猴数据预测患者血液学毒性的潜力。在该研究中,首先建立了抗体偶联MMAE(acMMAE)的PK模型,以预测食蟹猴和癌症患者的浓度时间曲线。采用acMMAE浓度,分别开发了临床前和临床semi-mechanistic中性粒细胞减少症模型。结果显示,可以用猴预测ADC的人体血液学毒性,以支持候选药物选择并制定临床风险缓解策略。

来源:药理毒理开发