
嘉峪检测网 2024-05-09 20:42
导读:本文介绍了常见的制药无菌工艺与如何评价无菌性。
无菌制剂的无菌性是确保用药安全的重要手段,这里介绍常见的制药无菌工艺,以及如何评价无菌性。
1、无菌工艺-Sterile和Aseptic区别
注射剂无菌保证工艺是指为实现规定的无菌保证水平所采取的经过充分验证后的灭菌(无菌)生产工艺。目前,注射剂的无菌保证工艺主要有两种:
终端灭菌工艺(terminal sterilization process), 一般来说,本方法成本低,无菌保证水平高,适宜于大容量注射剂的灭菌。
无菌生产工艺(aseptic processing) 是指以防止污染为目的,在无菌系统环境下,通过除菌过滤法或无菌操作法,消除导致污染的各种可能性来保证无菌水平。
无菌生产工艺和终端灭菌工艺具有不同的系统要求、不同的除菌方法和不同的无菌保证结果,这是由于无菌生产工艺对环境系统的要求高,且影响无菌操作的因素多而使得无菌保证水平比终端灭菌工艺低。
这里Sterile指灭菌,而Aseptic是无菌。前者代表更高的无菌保障水平。
2、无菌性Sterility
EMA定义:无菌性是指不存在活性微生物,其定义为无菌保证水平(Sterility Assurance Level, SAL)等于或小于10^-6。
欧洲药典:任何一个产品实现绝对的无菌,是不可能的,也无法证明。
美国药典:按照最严格的无菌定义,只有当物品中完全不存在活的微生物时,才能被称为是无菌的。
然而,这一书面定义无法应用于实际标识为无菌的物品,因为无菌检查的局限性无法消除,即: 不对每个物品均进行破坏性检查就无法证明物品绝对无菌。
使用无菌保障(Sterility Assurance)来表示对标识为无菌的物品安全性的确认。产品的无菌性以概率的方式定义。
3、无菌保障水平
Sterility Assurance Level
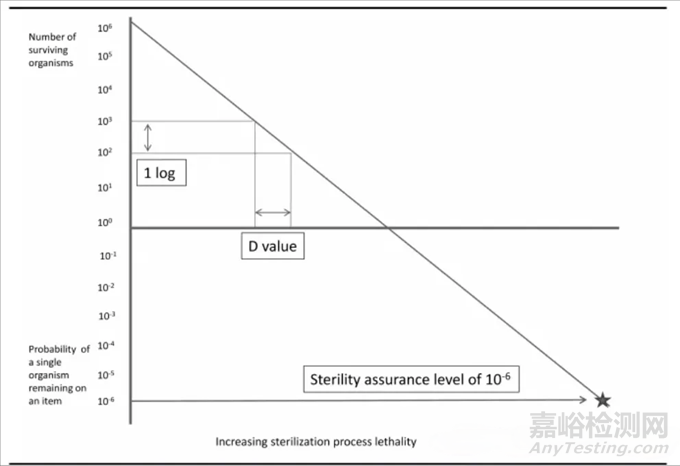
无菌保障水平,为产品经灭菌(除菌)后微生物残存的概率。SAL是评价灭菌(无菌)工艺的效果的重要指标。该值越小,表明产品中微生物存在的概率越小。
为了保证注射剂的无菌安全性,国际上一致规定,采用湿热灭菌法的SAL,不得大于10的(-6次方),即灭菌后微生物存活的概率不得大于百万分之一;而采用无菌生产工艺的产品,其SAL一般只能达到10的(-3次方),可见非终端灭菌制剂存在微生物的概率远远高于终端灭菌制剂,故仅限于临床必须注射给药而确实无法耐受终端灭菌的产品。
4、SAL测量和估算
无菌保障水平,为产品经灭菌( 除菌)后微生物残存的概率。可以采用生物指示剂来对其进行估算,通过杀灭一定孢子数的生物指示剂,例如>10^6个孢子数微生物。而实现10^-6,则需要根据生物指示剂特性,进行外推达到SAL≤10^-6。
估算方式,利用微生物在各种灭菌介质/条件下,微生物死亡速率符合或者近似一级动力学方程。其中微生物杀灭90%的时间为D值,是分析灭菌工艺效果的重要生物学参数。
D值(Decimal reduction time):就是在一定的处理环境中和在一定的热力致死温度条件下某细菌数群中每杀死90%原有残存活菌数时所需要的时间。
5、SAL来源和合理值
SAL的定义最早来源于食品行业,是瑞典国家卫生委员会于1965年提出,10^-6的无菌保障水平(SAL)。
据说该值是一个任意建立的质量标准,无任何临床相关性的质量标准。大家可以想想在医院静脉输液时,护士处理针头等的随意性,和SAL10^-6无法匹配。
需要根据不同的应用场景定义合理的无菌保障水平,达到微生物水平,满足产品特定的设计要求。例如终端灭菌的注射液要求SAL 10^-6,而热敏感的器械产品要求10^-3。
无菌保障对生物制品至关重要,不仅关乎产品质量、安全性和合规性,还关乎企业的声誉和市场竞争力。生物制品生产企业在生产过程中应高度重视无菌保障措施的落实。

来源:精密药事CF、CBioPC
关键词: 制药无菌工艺