
嘉峪检测网 2025-05-26 08:47
导读:本文介绍了干细胞药品的临床前药理毒理研究实验。
干细胞是一类具有自我更新和多向分化潜能的原始未分化细胞,它们在人体中扮演着重要的角色。干细胞能够分化成多种类型的细胞,包括但不限于血液细胞、神经细胞、肌肉细胞等具备替代和修复死亡和受损伤的细胞、免疫调节、激活休眠和处于抑制状态的细胞等作用,并可通过旁分泌作用,分泌多种生物活性因子,促进细胞增殖和抑制细胞凋亡。干细胞的这些功能使其在医学研究和治疗中具有巨大的潜力,特别是在组织修复、再生医学和免疫调节等领域。简单总结下干细胞的作用,就是“4R”,即repair(修复)、replace(取代)、restore(恢复)、regenerate(再生)。
目前用于细胞治疗产品的干细胞主要有4种:1)人胚胎干细胞(human embryonic stem cells, hESCs);2)诱导多能干细胞(induced pluripotent stem cells, iPSCs);3)成体干细胞(adult stem cells, ASCs)又称组织特异性干细胞或成体多能干细胞;4)基因编辑干细胞(gene-editing stem cells, GSCs)。hESCs是一种具有极高分化潜能的细胞,它们来源于早期胚胎(通常是受精卵发育到囊胚阶段的内细胞团)。hESCs具有发育全能性,理论上可以分化为成人体内的任何一种细胞类型。iPSCs是一种通过特定方法将成熟体细胞重新编程为具有多能性状态的干细胞。通过向成熟体细胞(如皮肤细胞或血液细胞)中引入特定的转录因子(如Oct4、Sox2、Klf4和c-Myc),可以将这些细胞转化为iPSCs。iPSCs具有与胚胎干细胞类似的多能性,能够分化成体内几乎所有类型的细胞。与胚胎干细胞相比,iPSCs的获取不涉及破坏胚胎,因此在伦理上更具优势。ASCs存在于成熟动物的许多组织中,负责维持和修复组织。通常与它们所在的组织或器官具有相同的特性,例如,造血干细胞主要存在于骨髓中,负责生成血细胞。与胚胎干细胞相比,成体干细胞的分化潜能通常有限。它们主要分化为所在组织的特定细胞类型,但某些成体干细胞也显示出跨系分化的潜力。GSCs是指利用基因编辑技术对干细胞的基因组进行精确修改的细胞。这种技术允许科学家在分子水平上改变或修复特定基因,以研究基因功能、治疗遗传性疾病或提高干细胞的医学应用潜力。
干细胞因其可自我更新、多向分化及旁分泌影响其它细胞群等特点,为非临床研究带来了很多挑战,结合公开文献和指导原则做下梳理。
干细胞产品的非临床评价需要回答以下几个核心问题。用于动物试验的干细胞,要进行充分的表征,包括但不限于细胞鉴定、纯度、活力、无菌和稳定性等。之后再通过动物模型、生物分布、毒理和成瘤性试验逐步表征干细胞的药理学和毒理学特点。

之所以要回答以上问题,很大程度上与干细胞这类产品的特点相关。所有的安全性风险来自于干细胞的增殖和分化特性。增殖和分化均有成瘤风险。一是有些增殖不可控,可能成瘤。二是有些非预期的分化会形成异位组织。最终分化的细胞(intended cell type)可能会进一步发育成熟后通过多种作用机制发挥作用,需要研究清楚。干细胞也有可能移行到远端组织,引起毒性。另外一个风险则是免疫原性,风险可能来自干细胞本身,也可能来自基因修饰的表达产物,涉及到免疫系统对受试干细胞的反应。非临床安全性研究很大程度上就为了评估这些风险。
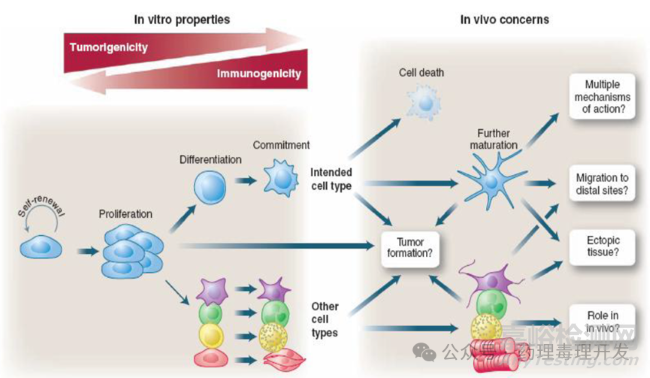
GLP依从性
原则上非临床安评应遵循GLP。如果是药效或生物分布中伴随安全性终点,也应最大限度按照GLP要求执行。
相关种属选择
首先,干细胞产品非临床研究没有默认种属,能提供所选择种属的科学依据即可。所选择的种属需是药理学敏感种属,即动物对人源干细胞及其分化后细胞的生物学反应与预期人体反应类似。为满足常规研究周期需要,动物要有3-12个月正常寿命。另外,动物试验系统需要是“progressive host”,允许细胞正常生存。FDA建议先在目标种属上开展预试验,看下接种细胞的生存情况。尤其拟采用免疫健全动物开展实验,如果免疫排斥明显,细胞不能正常长期存活,不适合在该种属开展正式毒理研究。
另外,所选动物尽量能满足相同的临床拟用装置和给药路径。大鼠(包括新生动物)、小鼠、非人灵长类、猪、手术动物、免疫缺陷动物模型等均有使用经验。大、小动物各有优劣,说不上谁好谁坏,大动物生理学和解剖结构与人体更为接近,方便采用临床拟用装置将药物递送到理想位置,并能保障充分的递送剂量。小动物则数量可以更多、更容易进行基因编辑,且有大量现成的免疫缺陷动物可用。
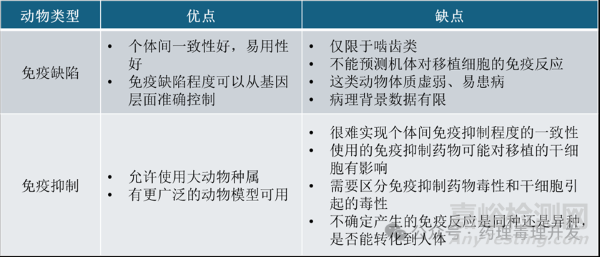
相比于健康动物,免疫缺陷动物潜在可减少免疫排斥,即免疫耐受性表现更好。免疫缺陷动物可以是先天缺陷,也可以给予免疫健全动物免疫抑制药物实现。免疫缺陷和免疫抑制动物模型也是各有优劣,如下表所示。
动物疾病模型有助于活性-毒性和风险-获益比的评估,也是推荐采用的。但动物疾病模型也是有优缺点的。优点包括:1)可以在病理生理状态下的局部微环境中评价产品的安全性;2)可以同时评估剂量/活性和剂量/毒性关系;3)鉴定潜在可用于指导临床的有效性和风险因素的biomarkers。缺点则包括:1)个体间变异大;2)缺乏可靠的历史背景数据和基线数据;3)潜在需要给予免疫抑制剂;4)动物护理问题和动物伦理问题。
其它因素如解剖学角度的器官和组织大小,是否允许使用靶向递送装置完成人体剂量递送,自体细胞的可获得性等也需要在种属选择过程中予以考虑。比如,用于人眼的RPE干细胞植入物体积较大,因此与人类眼睛大小相似的动物(如猪),是最合适的动物模型。
如果对干细胞进行了基因修饰,则被改造目的基因及其表达产物的种属相关性也需要考虑。
供试品
首选的自然是临床拟用产品。可以在免疫缺陷动物或者免疫抑制动物中开展。还有些干细胞在免疫健全动物中也可以开展非临床研究,因为有些接种位置是免疫豁免的(Immune privileged),或者有些细胞具备免疫豁免特点,不会被动物免疫系统排斥。
如果无法采用临床拟用产品,也可以考虑动物源替代产品,但要求比较高,无论生产工艺还是影响有效性和安全性的关键质量参数,均需要与人源产品进行比对。动物源产品的挑战包括可能产生与人源干细胞不同的生物学作用、缺少表征动物源干细胞的试剂、潜在产生不同的杂质。而且,不同种属间来源的干细胞之间的相似性研究尚不充分,缺乏统一的相似性评价标准。这类产品获得的数据,临床转化存在一定的不确定性。
另外,干细胞的包材和生物支持性材料的选择和表征,也很关键。
药理学研究
与其它传统药物药理学研究逻辑类似,也是分为体内和体外两部分,体外重点考察干细胞的表型和功能,如增殖、分化、分泌等能力,以及可能的基因改造引入的功能变化。比如有神经营养作用的干细胞需考察对细胞死亡的保护作用和/或分化成神经元的能力。对于潜在血管生成活性的内皮细胞,则要考察诱导产生血管结构的能力。体内则需要论证模型构建的科学性和合理性,并尽可能选择人体疾病相关动物模型开展。
药代动力学
在相关动物种属中考察干细胞药代行为,如生物分布、迁移、定植、增殖、分化、存续性等。生物分布可伴随药效学试验中考察。如果涉及基因修饰,对目的基因的存续、表达等也需要研究。
安全药理学
可结合在一般毒理学研究中进行。是否需要开展补充和追加的安全药理学研究,则视产品风险和情况而定,与其它药物的开展原则类似。
一般毒理学
种属
相关种属选择依据前文已有介绍。毒理试验原则上采用两种动物开展。如采用一种动物,需要提供科学合理性依据。通常采用双性别动物开展,如采用单一性别,也需要说明合理性。
剂量设计
同其他类型药物,一般设置多个剂量,如低、中、高三个剂量。高剂量可以药效学最高剂量的一定倍数、临床最高剂量的一定倍数或最大可行剂量。低和中剂量则在高剂量基础上设置合理剂间距确定。
试验设计
动物给药场景尽可能模拟临床,与临床用药场景尽可能接近(Mimic clinical scenario as closely as possible),包括细胞活力、浓度、制剂、体积、递送速率、植入或注射部位、植入或注射数量、递送系统、递送时间节点、递送方案、给药途径等。除了给药组和阴性对照组,可根据情况设置假手术组。
试验周期的长度取决于干细胞产品的体内存续时间和受试动物的寿命,短则4-12周,长则3-12个月。根据干细胞作用时间长短和给药次数,动物处理时间点一般设置为多点,可以分为时间点1(给药后48h或1周)、时间2(给药后2周或3个月)、时间点3(给药后3-4周或6个月)。
生物分布和毒理学方案简单设计如下表所示。有效性研究可以合并到毒理试验中。
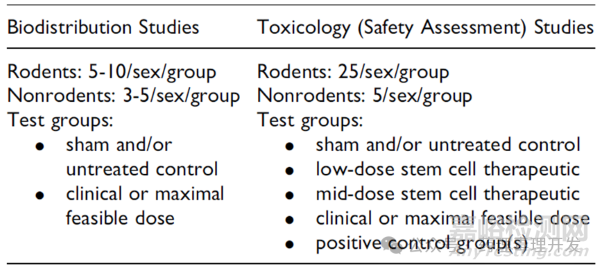
Q-PCR可以用于非靶组织分布的初筛,以减少IHC技术筛选非靶组织的需求。Q-PCR阳性的组织,可以再用IHC或者其它技术确认。通常干细胞毒理试验会设置多个动物处死时间点,考察急性、慢性和/或延迟毒性,及可能的毒性恢复情况。前序时间点,Q-PCR检测阴性,有助于减少后续时间点待检组织数量。如果对照组没有注射细胞,可以适当调整对照组组织收集的清单,仅采集必要组织。
检测方法
干细胞生物学作用比较复杂,对检测方法的要求很高。无论在人体还是动物模型中,经常会发现移植后干细胞的大量死亡,成功分化的数量比较有限。传统的病理HE染色可以检测到组织内数量比较多的干细胞,如果数量很少,HE就不那么适用了。这种情况下,需要对干细胞的作用机理有深入了解,干细胞发挥功能是通过直接发挥作用如组织再生,还是通过激活内源性干细胞或祖细胞发挥作用。可以通过一些biomarkers,采用诸如IHC、PCR、ISH的方法进行特异性评估。比如人源干细胞移植到动物体内,可以通过检测人核抗原、线粒体抗原监测人源成分。细胞增殖则可以通过增殖细胞核抗原(PCNA)或Ki67进行检测。
细胞支架
干细胞的制剂处方与传统生物药物也不同,为维持干细胞再生能力,制剂中通常会加入3D可生物降解支架、“支持性细胞”和/或其他生物活性分子,如骨形态发生蛋白(BMP)。多细胞混合组分使得安评更加复杂化,比如移植物抗宿主反应、支架材料的生物相容性、免疫原性等,需要重点关注。
试验终点
标准的安全性终点包括死亡率、临床观察、体重、体格检查、摄食量、饮水、临床病理(血生化、血液学、凝血、尿液分析)、器官重量、大体解剖和组织病理学检查等。基本与其它药物一般毒理学研究终点类似。
不过,干细胞由于不能被完全清除,一般不需要设置恢复期动物,但需要观察足够长时间,以评估慢性和脱靶毒性。
组织收集
与传统生物药物不同,病理学家在干细胞毒理评价中的作用尤为关键,不仅限于干细胞可能引起的毒理学变化,还可以通过评价干细胞形态、干细胞特定的表面抗原或可用于鉴别该细胞的biomarkers等,确定干细胞的组织分布、存续时间等。
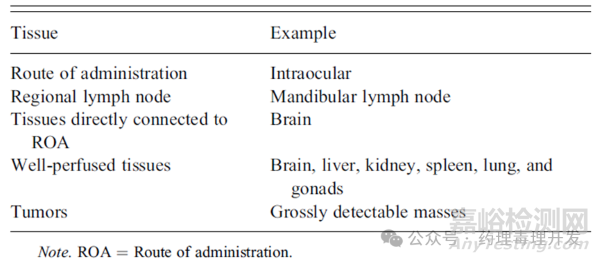
干细胞给药部位(route of administration, ROA)和附近的引流淋巴结与药效和安全性最相关。其次则是与给药部位直接相连接的组织。另外,血供最丰富的器官如肝脏、脾脏和肺也需要重点评价,可以评估干细胞随血液的移行扩散。再就是一些非正常的瘤块可能与干细胞的成瘤性相关。所以干细胞的组织病理评价可以根据风险进行分级,如下表示例。
成瘤性和致瘤性
未分化的干细胞有增殖上调的风险,可以分化或去分化成畸胎瘤或其它肿瘤,故成瘤性是干细胞产品的一个重要安全性风险。最好找到合适的biomarkers界定肿瘤是否来自人源干细胞。多能干细胞(如iPSCs和ESCs)的成瘤风险高于成体干细胞(如间充质干细胞、造血干细胞等)。
致瘤性风险指干细胞终产品促使正常细胞转变为肿瘤细胞的可能性。
对于成瘤性和致瘤性风险较低的成体干细胞产品,通常应在首次临床试验前至少完成体外成瘤性评价;对于拟用于非晚期肿瘤适应症且风险较低的干细胞产品,通常在上市前完成体内成瘤性和/或致瘤性试验。
对于成瘤性和/或致瘤性风险较高的干细胞产品(例如多能干细胞产品、其他分化程度较低的干细胞产品、药学和/或已有非临床研究提示风险较高的产品等),在首次临床试验前,应完成体外成瘤性评价、临床给药途径或敏感给药途径(如皮下注射、睾丸注射、肌腱注射、脊髓内注射等,应有合理依据)的体内成瘤性试验;若成瘤性试验出现阳性结果,或者一般毒理学试验中发现受者来源的癌前病变、可疑癌变等,首次临床试验前还应完成临床给药途径的致瘤性试验。若成瘤性试验结果为阴性,上市前完成临床给药途径的致瘤性试验。
遗传毒性
不需要开展标准组合的遗传毒性试验。如果设计基因修饰,需进行基因插入突变、基因编辑脱靶等风险评估。
免疫原性和免疫毒性、生殖毒性、制剂安全性
与其它药物类似,常规开展。免疫原性要考虑到动物是免疫缺陷动物还是免疫健全动物,根据产品特性和风险,考虑是否开展。如果涉及基因修饰,表达的目标蛋白的免疫原性也需要评价。
其它
FDA认为,干细胞试验可以考虑将POC+Tox+cell fate整合在一个动物模型中开展,更符合动物“3R”原则。Cell fate指的是细胞的存活、分化/表型、去分化、迁移、异位组织形成、增殖等变化。

来源:药理毒理开发
关键词: 干细胞药品