
嘉峪检测网 2024-07-11 09:59
导读:通过对已上市化学药品生产批量变更政策解读和山东省生产批量变更现状和常见问题进行分析,为相关单位开展生产批量变更研究工作提供参考。
摘要
目的 通过对已上市化学药品生产批量变更政策解读和山东省生产批量变更现状和常见问题进行分析,为相关单位开展生产批量变更研究工作提供参考。 方法 对《已上市化学药品药学变更技术指导原则(试行)》中批量变更章节进行深入解读,对山东省已上市化学药品生产批量变更现状和常见问题进行汇总分析。 结果 生产批量变更为《指导原则》实施后新增加的章节,且生产批量变更往往关联设备、工艺、参数、溶剂比例等生产过程中的各个方面,目前山东省生产批量变更的备案通过率有所改善,但申请人在对法规文件的理解上仍需提高。结论 新“两法”的实施,为申请人采用新技术、新设备,扩大产能,提高竞争优势提供了法规支持。 药品上市许可持有人作为药品上市后变更管理的责任主体,应当全面关注批量变更中的关联变更,更科学的开展药品上市后变更研究。
关键词:已上市化学药品;变更管理;批量变更;新法规
新修订《药品管理法》和《药品注册管理办法》(以下简称“新'两法’”)实施后,其配套文件《已上市化学药品药学变更研究技术指导原则(试行)(以下简称“《指导原则》”)首次增加了药品生产批量变更的相关要求[1]。虽然国内外已出台了连续制造的相关法规文件[2-3],该技术也引起了国内同仁的广泛关注[4-6],但在目前的国情下,传统批量生产技术仍然是制药行业生产的主流模式[6]。
《药品生产质量管理规范》(2010 年修订版)中“批”是经一个或若干加工过程生产的、具有预期均一质量和特性的一定数量的原辅料、包装材料或成品。为完成某些生产操作步骤,可能有必要将一批产品分成若干亚批,最终合并成为一个均一的批。在连续生产情况下,批必须与生产中具有预期均一特性的确定数量的产品相对应,批量可以是固定数量或固定时间段内生产的产品量。ICH Q7中对批的定义也有类似的描述。根据批生产的用途、功能等的不同,衍生出了实验室批、小试批、工艺验证批、临床批等不同的概念[7-8]。本文结合笔者工作实践,仅针对已上市化学药品的商业生产批的批量变更进行讨论,供相关研究者参考。
一、已上市化学药品生产批量变更的基本要求及分类
1.1 基本要求
生产批量变更是指在原批准批量(如关键临床试验批、BE批等)基础上扩大或缩小生产批量。原批准批量一般是指获批时的生产批量,如注册批件中注明的生产批量或生产工艺信息表中注明的生产批量。对于新“两法”实施前获批的品种,批件中一般没有明确批量信息,一般以当时注册申报的批量作为原批准批量;对于持有人或生产企业按照原生产工艺变更管理的有关规定和技术要求经研究、验证证明不影响药品质量的已实施的批量也可作为后续批量变更的基准。
《药物制剂人体生物利用度和生物等效性试验指导原则》对“受试药品”规定“试验用的受试药品应具有对将上市药品的代表性,例如,对于全身作用的口服固体制剂:①受试药品应来自一个不少于生产规模 1/10 的批次,或 100 000 单位,两者中选更多的,除非另外说明理由:②使用的生产批次应该确实保证产品和过程在工业规模可行。在生产批次规模小于 100 000 单位时,需要整个生产批次的样品供抽样用。”《指导原则》说明了生产批量缩小的情形下,当批量缩小至相关指导原则或技术要求规定的批量以下(如口服固体制剂批量缩小至 10万片以下)不在讨论范围内。同时因变更生产批量往往会引起工艺参数、生产设备等发生关联变更,若批量变更引发关联变更,需按照《指导原则》相关章节要求进行关联变更研究。
1.2 已上市化学原料药批量变更的分类及要求
《指导原则》中对已上市化学原料药批量变更分类比较明确:批量变更在原批准批量的10倍以内(包括 10 倍)属于微小变更,在原批准批量的 10倍以上的属于中等变更。需要注意的是化学原料药的批量变更往往关联生产设备、工艺参数甚至是生产工艺反应试剂、溶剂种类、合成路线等的变更,因此应关注关联变更的变更类别:若属于重大变更,应连同批量变更一并申报补充申请;若属于中等变更,应连同批量变更一并申报备案;若批量变更和关联变更均属于微小变更,申请人应做好相应的研究验证工作并在年报中进行报告。
1.3 已上市化学药品制剂批量变更的分类及要求
对于制剂的批量变更,《指导原则》按照口服固体制剂、非无菌半固体制剂、非无菌液体制剂、采用终端灭菌工艺的制剂、采用无菌生产工艺的无菌制剂以及特殊剂型制剂进行分类。
1.3.1 普通口服固体制剂和非无菌半固体制剂
普通口服固体制剂和非无菌半固体制剂的批量变更分类与原料药类似,在关键临床试验批或 BE 批批量的 10 倍及以内属于微小变更,10 倍以上属于中等变更。需要关注的是批量变更的微小变更,也可能会引起生产设备、工艺参数甚至是处方等的中等变更甚至是重大变更,因此要重点关注批量变更引起的关联变更的变更类别评估。普通口服固体制剂和非无菌半固体制剂基数较大,如截至 2022 年 4月,在已收录入《中国上市药品目录集》的 4 631个品规中,以口服固体制剂为主,其中片剂占一半以上;片剂、胶囊剂和颗粒剂3个剂型占总数的 65%左右[9]。山东省已上市化学药品生产批量变更事项中,片剂的批量变更就占到 35%左右,因此普通口服固体制剂和非无菌半固体制剂的批量变更是占比较大的事项,应引起足够的重视。
1.3.2 非无菌液体制剂
《指导原则》中非无菌液体制剂的变更均归属为微小变更,该事项同样需要注意的是批量的微小变更可能会引起生产设备、工艺参数等的中等变更甚至是重大变更,在申报时应将关联变更的研究验证一并申报,综合说明变更对产品质量的影响。
1.3.3 无菌制剂
《指导原则》中采用终端灭菌工艺的制剂,微生物负荷水平不变的前提下,批量变更按照溶液储存时间的增加来判定,不超过原批准时限的 50%时属于微小变更,超过 50%属于中等变更;采用无菌生产工艺的无菌制剂的批量变更,按照与无菌保障水平相关的步骤的生产时间(如配液,药液存放、过滤、灌装等)是否增加来判定,若上述时间增加属于中等变更。需要注意的是,上述是批量变更类别的判定,批量引起的关联变更的变更类别,如设备、工艺参数等,应按照《指导原则》的相关章节进行判断。
1.3.4 特殊剂型制剂
《指导原则》中规定,工艺复杂的特殊剂型制剂(如:缓控释制剂及肠溶制剂、透皮给药制剂、脂质体、长效制剂等)的批量变更属于重大变更,需要特别注意的是,对于工艺较简单的特殊剂型制剂,如凝胶骨架片或通过肠溶胶囊壳实现肠溶功能的肠溶胶囊[10] 批量变更对产品质量影响较小,可在充分的研究验证后通过沟通交流等形式实现降级管理。
二、山东省已上市化学药品生产批量变更的备案现状相关情况分析
2.1 山东省已上市化学药品生产批量变更的备案现状及不予备案情况
笔者对新法实施后,2021年至 2022 年山东省已上市化学药品生产批量变更的备案申请进行了数据统计,所有备案申请中,共有168 个生产批量的变更申请,其中制剂有 142个,通过一致性评价(包括视同通过一致性评价)的品种72 个,占制剂总数的 50.70%,具体见表1。
▲表1-山东省已上市化学药品生产批量变更备案情况及不予备案情况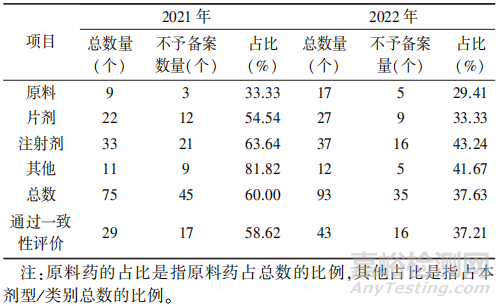
从上表可以看出,该事项2021年的不予备案率为60%,明显高于40%的总体不予备案率,这与批量变更是《指导原则》实施后新增加事项有关,申请人对法规的认识还有待提高。不论从单个剂型的备案成功率还是从过评品种的备案成功率来看,2022年比 2021年都有显著提高,但总体的不予备案率仍接近 40%,需要相关申请人继续提高对相关法规文件的认识。
2.2 山东省已上市化学药品生产批量变更品种纳入集采情况
药品的生产批量与产能直接挂钩,经笔者统计,168个变更批量备案申请中,有49个品种为纳人集采品种,占总数的 29.16%,占制剂总数的 34.5%以上,具体见表2。
▲表2-山东省已上市化学药品生产批量变更品种纳入集采情况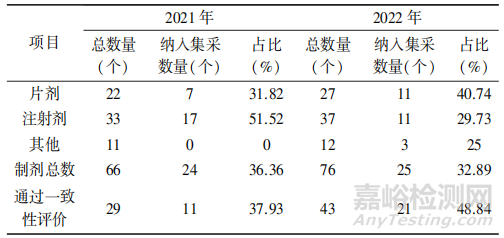
从上表可以看出,《指导原则》实施的第一年(2021年)纳人集采的产品批量变更集中在注射剂企业,第二年(2022年)批量变更品种集中在片剂,这与注射剂处方工艺相对简单,片剂处方工艺相对复杂有关;纳人集采的通过一致性评价品种批量变更占比较大,且有增加的趋势,新“两法”的实施,为申请人采用新技术、新设备,扩大产能,提高竞争优势提供了法规支持。
三、山东省已上市化学药品生产批量变更常见问题
因药品生产批量变更为新“两法”实施后《指导原则》中新增加的备案事项,批量变更不仅与产品生产过程相关,还可能引起生产设备、工艺参数、处方工艺等的变更,是《指导原则》中唯一一个微小变更也需要进行研究验证、提供批生产记录且进行溶出曲线对比的章节[10]。因其为新事项且相对复杂目前备案通过率不高,笔者统计的 2021 年至 2022年山东省已上市化学药品生产批量变更的168个备案申请中,有 80个品种不予备案,占 47.62%,申请人对该事项的理解程度和研究验证工作的认识程度都有待提高。笔者结合工作实践,对不予备案品种的常见问题进行了汇总,供相关研究者参考。
3.1 申请表备案的内容描述不清楚
申请表中第20 项“备案的内容”属于国家药监局官网备案信息公示时直接抓取的内容,对于多种规格、多个生产地址(线)的品种,可能存在不同规格、不同生产地址(线)生产批量不一致的情况,在申报时应将相关信息描述清楚,明确变更的具体品种、规格以及生产线等信息,这样才不会在公示内容中出现错误。
3.2 未对关联变更进行同步研究申报
目前90%以上批量变更的申请会发生生产设备、工艺参数甚至会引起原料药反应试剂、溶剂种类和比例、生产工艺等关联变更。若申请人未对关联变更进行相应的研究、申报,变更类别可能判断的不准确。如某原料药品种批量变更为12倍,按照指导原则关于批量变更的规定属于中等变更;但其在最后一步反应后使用了不同设计/原理的生产设备,且最后一步反应后有生产工艺的关联变更,这种情况按照中等变更申报备案显然是不符合《指导原则》要求的。
3.3 通过一致性评价品种未提供生产工艺信息表
对于通过一致性评价品种,生产工艺信息表会提供生产工艺、设备、参数、标准和中间体控制等相关信息,是备案审查的重要依据,也是品种变更的基础。申请人应将产品的生产工艺信息表作为重要文件,对比变更前后工艺、设备、参数和中间体控制等是否发生变更。比如,口服固体制剂的批量一般是以最终总混的样品为准计算,申请人经常是以增加亚批的形式扩大批量,此种情况工艺研究时,应充分考察亚批之间的质量一致性[11]。产品批量放大,还应充分考察中间过程控制指标的变化,有效避免或控制其中的质量影响因素,才能确保产品质量可控[12]。若申请人未提供生产工艺信息表,则无法对比变更前后的亚批情况、亚批之间一致性以及中间体控制情况等的一致性。
3.4 变更前批量确定不符合要求
《指导原则》中规定生产批量变更是指在原批准批量基础上扩大或缩小生产批量,部分申请人将前期发生过微小变更后的生产批量作为后续批量变更的基准,显然是不符合指导原则规定的。例如,某普通口服片剂原批准批量为10万片/ 批,申请人前期通过微小变更将批量变更为50万片/ 批,后续拟将批量继续扩大为150万片/ 批,应属于在原批准批量基础上扩大 15倍的中等变更情形,而不是以50万片/ 批为基础扩大3倍的微小变更情形。
3.5 无菌产品验证资料不全
无菌产品验证资料不全,是无菌产品批量变更中的常见问题。无菌产品的灭(除)菌工艺是保证制剂质量和用药安全的关键步骤。无菌制剂的批量变更一般是与生产时限的变更同步的,对于无菌生产工艺的无菌制剂,需要关注无菌工艺验证的相关数据是否支持生产时限的变更;对于采用终端灭菌工艺的无菌制剂,应关注溶液储存时间变化前后微生物负荷水平是否发生变化。同时需要注意的是,无菌制剂的批量变更往往会引起灭菌工艺/冻干工艺中装载方式的变化,申请人需针对装载方式的变化对产品质量的影响进行相应的研究验证工作。
3.6 质量/稳定性对比研究不全面
批量变更是《指导原则》中唯一一个微小变更也需要提供溶出曲线对比数据的章节,并且也是为数不多的微小变更也需要进行质量对比的章节,该事项质量/ 稳定性对比不全面的问题主要表现在:①未对变更生产批量前后的样品进行质量对比研究或质量对比研究项目不全(如缺少有关物质检查项);②未进行溶出曲线对比;③稳定性对比中无有关物质项;④稳定性考察项目不全面,如:未考察失水率等;⑤稳定性考察条件不符合相关指导原则的要求等。上述问题不符合现行的技术要求,不能全面、客观地反应产品质量的变化[13-14],无法保证变更前后质量的一致性。
四、结语
新“两法”的实施,使药品上市后变更的法律法规体系更加完善,相关变更程序也发生了很大的变化,药品生产批量的变更常常关联设备、工艺、参数、溶剂比例等生产过程中的各个方面,笔者结合工作实践和相关文献,对《指导原则》批量变更章节中相关分类进行解读,并结合审评实践介绍了批量变更备案的现状以及常见问题,供相关研究者参考。同时,持有人应发挥药品上市后变更管理的主体责任,应当全面关注批量变更中的关联变更[10],主动按照相关法规、技术指导原则的要求,更科学地开展药品上市后研究,实现全生命周期管理。
参考文献
[1]张保梅,陆骁骏.结合中美欧的上市后变更管理制度浅谈我国新版上市后化药变更指导原则[J].中国医药工业杂志,2022,53(7):1056-1061.
[2]国家药品监督管理局药品审评中心.《已上市化学药品药学变更研究技术指导原则(试行)》的通告(2021年第15号)[EB/OL].(2021-02-10)[2023-06-20].https://www.cde.org.cn/main/news/viewInfoCommon/4ec3dca752a82B47bdf24ad3d3e85113.
[3]ICH.Q13:Continuous Manufacturing of Drug Substances andDrug products,Step 2 Document to be Released for Comments[EB/OL].(2021-08-12)[2023-0620].https://database.ich. org/sites/default/files/Q13Step2/Presentation20211004.pdf.
[4]] 孙钟毓,林泊然,李爽爽,等.国内外已上市连续制造口服固体制剂药学审评内容的研究与启示[J].中国食品药品监管,2022(9):54-77.
[5] 曹辉,丁力承,胡延臣,等.国外已上市连续制造药品的注册审评情况及其启[J].中国药事,2022,36(4):377 -383.
[6]曹萌,葛渊源,胡延臣,等.口服固体制剂连续制造物料处理考虑及监管检查要点探讨[J].中国医药工业杂志,2022,53(10):1394-1401.
[7]任连杰,刘涓,马玉楠,等.仿制药申请注册批次生产规模的思考[J].药学进展,2017,41(10):783-787.
[8]王宏亮,陈震.关于化学药品注册批量问题的探讨[J].中国新药杂志,2016,25(18):2046-2051.
[9] 周冲,武海军,王爱竹.凝胶类药品仿制药一致性评价药学研发要求和参比制剂分析[J].中国食品药品监管,2023(2):58-63.
[10]王淑华,陆骁骏,陈爱萍,等.《已上市化学药品药学变更研究技术指导原则(试行)》制剂生产工艺、批量及包材变更的解读[J].中国食品药品监管,2022(8):46 -51.
[11]石靖,许真玉.口服固体制剂一致性评价处方工艺研究的常见问题分析[J].中国新药杂志,2019,28(13):561 -1566.
[12] 章俊麟,石勇平,那馨竹,等.化学药品特殊注射剂仿制药药学研究技术要求[J].中国新药杂志,2023,32(2):63-167.
[13] 王筱婧,王东兴,祝倩,等.药品技术转让质量对比研究的主要内容与关注要点[J].中国医药科学,2016,6(14):221-223.
[14]周冲,董爱梅,刘军田.山东省化学药品上市许可持有人(试点)委托生产质量研究常见问题分析[J].食品与药品,2020,22(4):290-293.

来源:药学研究
关键词: 化学药品