
嘉峪检测网 2025-01-03 17:45
导读:本文将对灭菌追加确认步骤进行介绍,给需要进行产品追加确认的企业提供参考。
环氧乙烷灭菌是无菌医疗器械产品常用的一种灭菌方法。在形成常规灭菌程序前首先应进行环氧乙烷灭菌过程确认。而产品追加是将待选产品引入现有已确认的环氧乙烷处理组或环氧乙烷产品族的过程。使用产品追加确认的方式可以有效的节约企业成本,但如何进行有效而充分的产品追加确认却是困扰很多企业的难题。本文将对灭菌追加确认步骤进行介绍,给需要进行产品追加确认的企业提供参考。
第一部分:识别待选产品和当前已确认产品的差异
如采用产品追加确认的方式将待选产品纳入已有的环氧乙烷处理组或环氧乙烷产品族,应参照标准YY/T 1268 《环氧乙烷灭菌的产品追加和过程》,开展环氧乙烷追加研究并提供评估报告。本文里提到的待选产品为新产品或有变化的产品(改进的产品),而进行追加确认的过程实质就是对比待选产品与已确认的产品或已确认的过程挑战装置对灭菌过程的相对抗力的过程。
按YY/T 1268 《环氧乙烷灭菌的产品追加和过程》的要求,首先要做的就是识别待选产品和当前已确认产品的差异并确定已识别差异的潜在影响,这些影响确定包括但不限于以下几个方面:
1. 对产品不良影响的确定;
2. 对产品设计确定的影响;
3. 对材料和特性影响的确定;
4. 对无菌屏障系统影响的确定;
5. 对装载结构影响的确定。
第二部分:产品追加需要回答的问题
为了更好的识别待选产品和当前已确认产品的差异,在识别的过程中需要按标准YY/T 1268 《环氧乙烷灭菌的产品追加和过程》中附录A产品追加至环氧乙烷产品族或环氧乙烷处理组的评估指南,回答以下4大方面的40个问题。若问题中任一问题的答案为“是”,则要求对待选产品的产品性能研究进行进一步评估以确定待选产品是否比现有已确认产品或过程挑战装置更难灭菌。
1. 设计特征
与已确认产品相比,待选产品是否有:
a)更受限的通道或内腔;
b)更少的开口;
c)更多的内表面;
d)更多的结合面;
e)更多的闭合。
2. 材料及其他特征
与已确认产品相比,待选产品是否有:
a)材料或特性变化或差异,可能减少热量、湿气或灭菌剂穿透;
b)已知能吸附更多环氧乙烷残留量的材料;
c)会被环氧乙烷破坏的材料;
d)更多生物源性的材料;
e)现有环氧乙烷过程无法满足的温度、压力或湿度限制;
f)在型号、数量和抗力有重大不同的生物负载;
g)不受控环境下发生的制造或装配过程;
h)更少的过程清洁;
i)生产过程中更多的人工处理。
3. 无菌屏障系统
与已确认产品相比:
a)任何如产品、产品包装或不渗透纸是否使通气口变得更阻塞?
b)与现有包装在类型、层数、克重、涂层或处理方式是否有不同?
c)透气材料类型是否有不同(如用透析纸代替无纺布)?
d)透气材料的多孔性是否有降低(如基重、涂层、处理方式或粘贴标签的应用)?
e)透气材料的表面积或基础开口是否有減少?
f)包装材料使产品生物负载水平是否有增加?
g)是否增加了第二层(双层)无菌屏障系统?
h)包装设计、材料或产品的放置是否使产品更难加热或限制了气体在产品内流动?
i)灭菌过程是否对包装材料或封口有不良影响?
j)是否由于其他产品或自身的保护性包装的不透气性,使保护性包装内单包装的排列导致透气口变的更闭塞?
4. 装载特性
与已确认产品相比:
a)外包装是否有增加或变化,或内部隔层包装在数量上是否有增加?
b)是否增加了额外的保护性包装?
c)在处理过程中,为固定托盘装载是否使用了更厚或密度更大的伸展或收缩包装?
d)保护性包装材料的成分、密度或厚度有变化吗?
e)保护性包装材料是否有任何增加或变化使产品更难加热,或减少了环氧乙烷、湿气和空气的流动或扩散,或影响环氧乙烷残留?
f)“使用说明书”的放置或结构是否影响包装系统?
g)放置待选产品的整个托盘或装载的密度是否有变化且该变化已超过已确认的密度范围?
h)托盘装载结构是否更密实或暴露箱体表面更少?
i)在托盘内使用的气体通路或其他气体空间是否减少了?
j)柜室内的总装载量是否增加或减少了?
k)在结构上是否有减少热传输或气流的变化且该变化能影响产品环氧乙烷残留?
第三部分:产品追加确认流程
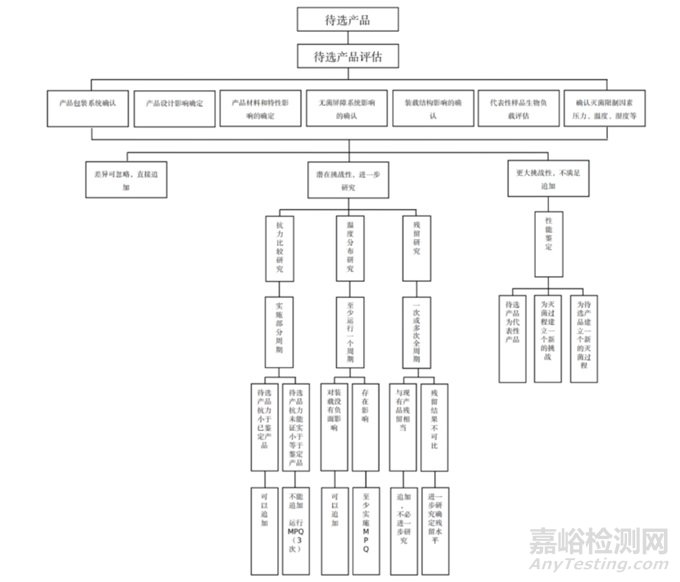
第四部分:产品追加确认文件输出
产品追加确认过程中需要输出的文件:
1. 追加产品在进行放行时所实施的进一步的测试:如残留测试、功能性测试。
2. 分析结果形成文件包含内容:
a)待选产品规格,灭菌过程中呈现的包装和装载结构,以及所需的无菌保障水平;
b)待选产品和现有已经确认产品的比较结果(应清晰的评估了产品的复杂性、材料、包装和装载结构),也应包括评审评估以保证待选产品功能未因追加至该过程而受到影响;
c)待选产品的生物负载以及生物负载与过程挑战装置的相对抗性证据或评估,
d)待选产品环氧乙烷残留水平的评估;
e)追加后产品进行常规放行的确认或鉴定实施的进一步测试等,如残留,功能,性能等测试;
f)灭菌专家和组织内实施变更的所需的其他人的批准。
第五部分:常见问题及应对措施
在产品申报注册或进行注册核查过程中,产品的追加确认通常会遇到以下三种结果:
1. 待追加产品与已经确认产品/产品族或处理组产品相似或细微的差异可忽略,可以直接导入。
2. 待追加与已经确认产品/产品族或处理组产品相比更具潜在的挑战性(灭活挑战性或热力挑战性或残留方面的挑战性) ,需要进一步研究确认待追加/产品族产品是否可以追加到现有已确认的灭菌工艺中。
3. 待追加与已经确认产品/产品族或处理组产品相比差异显著,无法进行追加。
针对以上三种结果推荐以下应对措施:
1. 待追加产品与已经确认产品/产品族或处理组产品相似或细微的差异可忽略的情形
√ 实施待选产品生物负载测试以证明与过程挑战装置相对抗性评审提供支持性证据。
√ 实施至少一个已经确认灭菌工艺的全周期(可以在日常常规参数下进行)以评估灭菌工艺与产品功能等相适宜性。
√ 在全周期中通常进行产品功能测试、残留测试等测试。
2. 待追加与已经确认产品/产品族或处理组产品相比更具潜在的挑战性(灭活挑战性或热力挑战性或残留方面的挑战性) ,需要进一步研究确认待追加/产品族产品是否可以追加到现有已确认的灭菌工艺中。
√ 实施待选产品生物负载测试以证明与过程挑战装置相对抗性评审提供支持性证据。
√ 实施一个部分周期以评估待选产品与已确认过程挑战装置的抗性。
√ 实施至少一个已经确认灭菌工艺的全周期,以评估灭菌过程中热力挑战性或残留方面的挑战性以及灭菌工艺与产品功能等相适宜性。在全周期中通常进行产品功能测试、残留测试,以及其它定义(无菌屏障/包装测试)等测试。
3. 待追加与已经确认产品/产品族或处理组产品相比差异显著,或按照2中进一步研究后,无法进行追加的情形,则对待追加产品/产品族产品依据标准GB 18279.1 《医疗保健产品灭菌 环氧乙烷 第1部分:医疗器械灭菌过程的开发、确认和常规控制的要求》,进行完整的性能确认。
第六部分:术语
1. 产品追加(productadoption):将待选产品引入现有已确认的环氧乙烷处理组或环氧乙烷产品族的过程。
2. 待选产品(candidateproduct):拟追加至现有已确认的灭菌过程中的新的或变更过的产品(含包装系统)。
3. 环氧乙烷处理组(EOprocessingcategory):能在同一环氧乙烷灭菌过程中灭菌的不同产品或产品族的组合。
注:处理组内所有产品呈现对灭菌过程的挑战性不大于处理组的挑战装置。
4. 环氧乙烷产品族(EOproductfamily):允许采用相同的工艺条件进行灭菌并具有相似或相同确认目的的产品组合。
5. 过程挑战装置(processchallengedevice;PCD):对某一灭菌过程构成特定抗力的装置,用于评价该灭菌过程的性能。
注1:本文件中过程挑战装置可以是产品、模拟产品或直接/间接接种的其他装置。
注2:本文件区分了内部过程挑战装置和外部过程挑战装置。内部过程挑战装置用于证明要求的产品无菌保证水平已达到。放置于产品内部或销售包装内的过程挑战装置是一个内部的过程挑战装置,而放置于产品销售包装之间或装载的外表面上的过程挑战装置是外部过程挑战装置。外部过程挑战装置用于常规灭菌周期的微生物监视。
6. 装载结构(loadconfiguration):产品在灭菌过程中表现形式的总属性,此结构包括:
a) 产品在无菌屏障系统(初包装)内的方向;
b) 无菌屏障系统在保护性包装(第二层或第三层包装)内的数量和方向;
c) 在灭菌柜托盘上(或搬运器内),产品在保护性包装内的数量、方向和放置方式;
d) 托盘(或搬运器)在灭菌柜或灭菌区域内的数量和放置方式。
7. 无菌屏障系统(sterilebarriersystem):为了产品在使用时保持无菌,防止微生物进入的最低限度的包装。
8. 包装系统(packagingsystem):无菌屏障系统和保护性包装的组合。
9. 灭菌专家(sterilizationspecialist):掌握应用的灭菌技术以及该灭菌技术对材料和微生物影响知识的人员。
注:通过实践和理论两种方式获得该灭菌知识水平,上述人员不需要所涉及的相关灭菌技术基本原理的指导。
10. 产品族(productfamily):具有某些特性,允许使用同一已定义的过程条件灭菌的产品系列。

来源:北京药监局