
嘉峪检测网 2025-04-24 09:19
导读:文章阐述了实施参数放行的灭菌系统中,确保所有关键参数和重要参数始终如一地符合预定要求的要素及其评价策略。
摘要
参数放行是指用基于对关键工艺控制参数的审核替代依赖最终产品的检验,放行最终灭菌产品的无菌放行程序。文章阐述了实施参数放行的灭菌系统中,确保所有关键参数和重要参数始终如一地符合预定要求的要素及其评价策略,包括灭菌系统设计的回顾与研究、灭菌温度监控、装载方式、灭菌程序运行参数、二次污染、产品隔离、人员。有效实施参数放行,有利于企业深化对产品工艺的理解和控制,进一步提升企业的质量管理水平。
【关键词 】参数放行 ;最终灭菌产品 ;灭菌系统 ;关键要素 ;无菌保证水平
参数放行 (parametric release),是指在严格实施 GMP 的基础上监控生产全过程,基于关键参数符合既定标准,对产品的无菌保证进行评价,以确保达到 ChP 2020 年版规定的无菌保证水平 (sterility assurance level,SAL) 不高于 10–6,从而替代最终产品无菌检查的放行系统 [1]。实施参数放行的前提条件是相关灭菌工艺经过充分验证,且在灭菌程序运行过程中灭菌工艺始终受到有效控制和监测,并有完整的文档记录。
灭菌系统作为药品生产过程中保障产品无菌保证水平的关键系统,确保其受控、有效、稳定运行是实施参数放行的重点。本文将讨论最终湿热灭菌药品实施参数放行对灭菌系统的特别要求和实施措施。
1.灭菌系统的关键要素
实施参数放行,必须要以良好的质量管理系统为基础。针对灭菌系统,需充分理解和掌握有关湿热灭菌的科学原理,这是正确实施参数放行程序的必要前提条件。此外,还应了解影响灭菌效果及保持无菌特性的潜在因素。本节主要阐述在实施参数放行的灭菌系统中,可确保灭菌效果的重要或关键参数始终符合预定要求的要素。
1.1 灭菌系统设计的回顾与研究
作为实施参数放行的重点以及确保产品 SAL 的关键,灭菌系统的设计与研究是确保其有效且稳定运行的基础。
1.2 灭菌温度
对于湿热灭菌系统而言,判断灭菌器性能的关键指标是温度。灭菌系统中通常装载探头,可直接测得容器内的温度。因此,使用装载探头测得的数据被视为关键参数 [2]。当测得数据符合灭菌工艺要求时,说明产品的 SAL 能够得到保障。
1.3 装载方式
液体产品灭菌时,灭菌介质的能量是通过容器传递的。任何情况下都应考虑加热对液体的影响,因此必须明确装载方式 [3]。液体产品的包装容器类型、灌装体积等差异,可能影响温度分布、热穿透性能,甚至杀灭效果。因此,需根据产品特性确定其装载方式。
1.4 灭菌程序运行参数
产品在特定的装载方式下,为确保能始终获得足够的物理 / 生物灭菌效果,同时又确保产品的包装能够保持完整、产品组分和效价不发生改变,在灭菌工艺开发阶段需确定灭菌程序的运行参数。运行参数是评价产品能否实施参数放行的关键。
1.5 二次污染
为了避免灭菌后产品发生二次污染,需控制和定期监测灭菌用水 ( 加热或冷却用水 ) 的微生物水平 [4]。用于灭菌腔室内过压或真空解除的气体 ( 通常是空气或氮气 ),不得对灭菌产品造成污染或二次污染。
1.6 产品隔离
应防止已灭菌产品与未灭菌产品混淆。可采用物理隔离措施降低产品混淆的风险,或采用化学指示剂、扫码追溯的方式区分已灭菌与未灭菌产品,并通过有效的管理规程强化对产品的隔离。
1.7 人员
生产现场灭菌岗位和质量保证部门人员的专业能力和工作素养,决定了生产时灭菌工艺是否得到有效监测和控制。设备使用与维护人员的专业能力和工作素养,决定了灭菌系统是否能够持续稳定运行。
2.灭菌系统的评估
灭菌系统能否有效、稳定地运行,并持续确保产品的灭菌效果,需要企业对灭菌系统的控制水平进行评估。实施参数放行的企业,应建立完善的质量风险管理系统和规程,涵盖影响产品无菌保证的所有方面 ;建立以质量风险管理为基础的无菌保证系统 ;设计并验证灭菌工艺,充分评估并消除放行不符合参数标准产品的风险。
企业可采用风险分析工具来识别风险并采取纠正和预防措施,利用风险评估对灭菌系统的关键要素进行评估。
2.1 灭菌系统设计的回顾与研究
企业应对灭菌系统的设计进行回顾性风险评估。灭菌系统通常包括灭菌器、监测和控制系统,以及为灭菌器提供支持的公用介质系统。针对现有产品或工艺实施参数放行,灭菌系统的风险评估应考虑对历史数据的回顾分析,包括灭菌工艺验证历史、灭菌设备验证历史、日常灭菌数据等 [5]。
灭菌器作为实施参数放行的关键设备,其性能保证尤为重要。灭菌器性能是否达到参数放行的要求,应基于灭菌器的设计和制造进行判断,并通过验证活动加以证实。灭菌监测装置 ( 如温度传感器、压力传感器 ) 应能监测每个循环的灭菌过程。应制定校准监测装置的操作规程及计量器具的可接受标准。计量器具的校准可追溯至国家标准或国家认可的国际标准。
针对隐性参数的研究尤为重要。因为隐性参数发生变化时,难以从关键参数反映出来,但对灭菌效果却存在较大风险,例如喷淋盘安装水平度改变、喷嘴堵塞等情况。针对隐性参数的研究,企业可以通过人为制造异常情况来开展。例如,研究喷淋盘安装水平度对温度的影响时,可人为地将喷淋盘紧固螺丝松开,模拟运行过程中喷淋盘倾斜的状况,以测试该情形下温度的变化趋势。我司实践中观察到,在体积为 36 m3 的灭菌器中进行灭菌,如果喷淋盘一边倾斜 1 cm,将导致该区域的产品温度偏低0.4 ℃。因此,只有对隐性参数进行充分研究,才能在异常情况发生时依据研究数据快速找出故障原因,并进行相应调整。
2.2 灭菌温度监控
灭菌温度是灭菌系统的关键参数,应确保灭菌器腔室内的温度被准确控制在规定范围内。通常需要配置独立的双芯探头来测定腔室内温度,探头的两芯应并肩排列在一起,同时测定灭菌期间的温度并将信号分别传送到灭菌控制系统和监控系统。在灭菌保温阶段,温差应控制在规定的限度范围 ( 通常为校准系统误差 ) 内,从而确保并验证灭菌过程中的温度受到控制。另外,两芯测得的温差应在规定的限度范围内,从而保证探头测定灭菌过程温度的准确性。
目前,多数企业采用无纸 / 有纸记录仪作为灭菌监控系统,但该方式无法实现温度探头两芯温度的自动对比。建议将记录仪更换为监控系统,并与控制系统联动,从而实现两芯温度的自动、实时比对 ;同时,应设置报警值,当两芯的温差超出规定范围时自动报警,以利于实施参数放行。
虽然装载探头能直接测得容器中的温度,通过控制系统自动计算出 F0 值 (F0 值为标准灭菌时间,即灭菌过程赋予被灭菌物品 121 ℃下的等效灭菌时间 ),以此直观反映被灭菌产品的无菌保证水平,但该方式仍具有一定的挑战性。装载探头的数量通常有限,只能测定灭菌器腔室中部分位点的温度,这样得到的检测结果能否反映每个产品的灭菌状况,是企业面临的最大挑战。因此,在产品的灭菌程序中,装载探头的位置必须具有代表性,应通过验证试验证明该设计能达到灭菌工艺的要求,确保被灭菌产品的 SAL。例如,应考察探头插入产品的位置是否为单个容器内的冷点位置,探头所测温度与被灭菌产品温度的对应关系等。
下面以直立式聚丙烯输液袋包装产品单个容器内的冷点确认,以及包装容器干点灭菌效果确认为例,介绍相关内容。
2.2.1 直立式聚丙烯输液袋包装产品单个容器内的冷点确认
根据《PDA.TR1 湿热灭菌验证》(PDA Technical Report No.1,Validation of Moist Heat Sterilization Processes:Cycle Design,Development,Qualification and Ongoing Control)[6]中的描述,“对于大容量注射剂而言,冷点位于产品几何中心和纵轴的底部。”以规格为 100 mL 的直立式聚丙烯输液袋包装产品为例,开展单个容器内冷点确认研究。选取产品上部、中部、底部进行容器内温度测试 (115 ℃,维持30 min),确认产品容器内冷点。测试结果显示,上部、中部、底部的平均温差最大为 0.07 ℃,F0 值的差值最大为 0.67( 图 1)。

▲图1-大容量输液袋中不同位置液体的F0值
该结果证明,直立式聚丙烯输液袋包装产品容器内冷点位于几何中心和纵轴的底部。当采用直立方式装载产品并通过顶部喷淋盘喷淋的水浴式灭菌器进行灭菌时,喷淋水由上至下对产品进行淋浴加热,由于热对流因素的影响,上层液体达到灭菌温度的速度要快于中层和下层液体,且产品规格越大,该差异越明显。
2.2.2 包装容器干点灭菌效果确认
根据《PDA.TR1 湿热灭菌验证》[6]中的观点,注射剂包装容器存在干点,即包装容器中最难穿透的点 ( 始终未接触液体的部位 )。灭菌应保证包装容器的干点位置也能达到灭菌效果,即达到 SAL 小于 10–6 的水平。
通过分析聚丙烯输液瓶注射液的包装容器特点,可见其组合盖内盖的上下部位为包装容器干点。根据该形状特点设计试验,并进行生物学确认。将含嗜热脂肪地芽孢杆菌的菌片放至组合盖垫片的上、下部位 ( 如图 2 所示 ),然后组合、焊盖,再进行灭菌。将如上制备的聚丙烯输液瓶放于灭菌器的冷点位置进行灭菌,连续 3 次 ( 灭菌工艺为 121 ℃、12 min)。结果显示,连续 3 次的组合盖内部灭菌试验中所有已灭菌生物指示剂均被完全杀灭,有效证明了该灭菌工艺条件下可保证包装容器干点的灭菌效果。
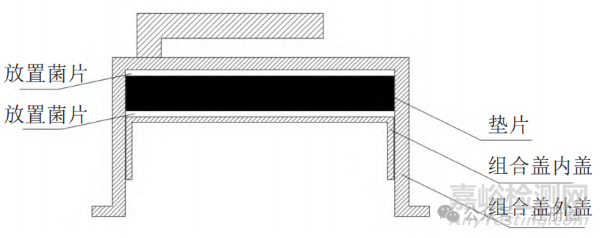
▲图2-菌片放置示意图
2.3 装载方式
各灭菌程序和各装载方式需进行热分布和热穿透试验。装载方式的开发最初可在研发环境下进行,所用灭菌设备应与生产用灭菌器采用相同或采用相似的程序条件,包括装载方式、灭菌时间和温度、压力以及热能的输入值 (F 值 )。装载方式的开发也可在生产现场进行。
现有产品实施参数放行,其装载方式需评估灭菌器装载区域内产品的定位、堆叠和分布,包括每种装载被送入灭菌器过程以及灭菌过程中发生损坏的可能性,此外还应包括由于上述因素导致产品无法达到无菌的风险。另外,应评价最大和最小装载是否是对该灭菌程序下灭菌效果最具挑战性的装载方式,从而为制定验证方案提供依据。
2.4 灭菌程序运行参数
灭菌保温时间、压力和温度对每个灭菌程序至关重要。为确保灭菌程序稳定运行,每个运行参数会有 1 个设定值,并且有一定的允许波动范围。波动范围的大小取决于灭菌工艺的运行能力、校验的允许误差和参数对灭菌效果的影响程度。在确定波动范围时,应考虑在灭菌的下限能否保证产品的 SAL符合要求,上限能否保证产品在有效期内的完整性( 包括包装密封性以及产品组分的降解产物符合经批准的限度标准 )[7]。《PDA.TR1 湿热灭菌验证》[6]中介绍了程序设计法,确定了灭菌程序所要具备的物理 / 生物杀灭效果。当杀灭效果被确定后,需要对可接受程序条件范围的下限进行多次运行和测试,以确保所设定的程序能持续、稳定地赋予被灭菌物品所要求的杀灭效果。
灭菌程序的运行参数分为关键参数和重要参数 [8]。①关键参数,指在灭菌过程中既受到控制又受到监测并且直接影响灭菌效果的数据。任一关键参数不合格,将导致产品被拒绝放行 [9]。保温时间、压力和温度或 F0 值为关键参数。一旦出现关键参数不合格,必须进行调查并采取纠偏措施,以防再次发生。②重要参数,指对保证灭菌程序运行状态稳定起重要作用的参数。重要参数应被监测或控制。任一重要参数不合格,需要调查原因,并书面说明相关偏差对相关灭菌程序灭菌效果的影响程度。灭菌时的加热和冷却时间往往是典型的重要参数。
企业可开展关键参数和重要参数的相关研究,进一步确定运行时可接受的参数范围。以蒸汽对温度影响的研究为例,确定产品可接受的灭菌蒸汽压力范围。
在蒸汽可供应的压力上限和下限值情况下开展温度测试。选用 500 mL 软袋产品,以湿热灭菌工艺 (121 ℃、12 min) 灭菌,测定蒸汽压力上限(0.6 MPa) 和蒸汽压力下限 (0.3 MPa) 条件下的升温时间、温度均匀性、F0 值,测试结果如图 3 所示。
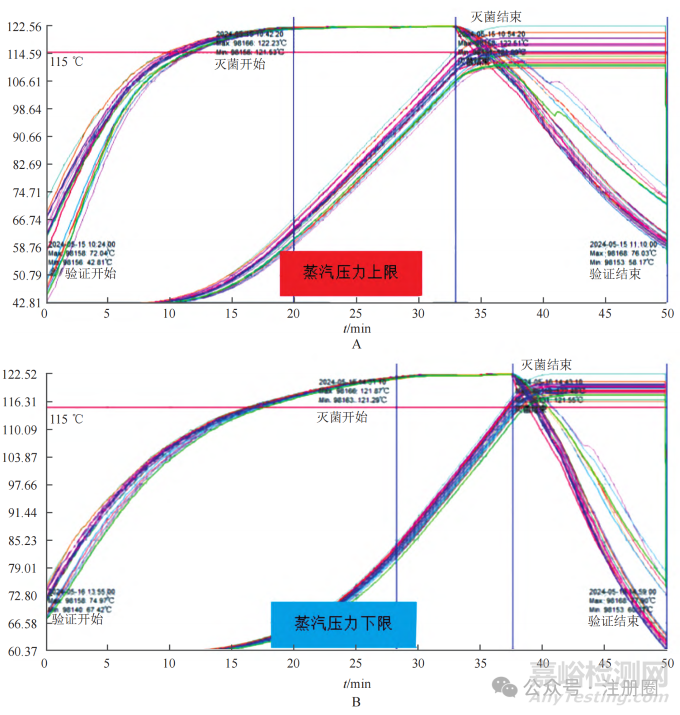
A—蒸汽压力上限 ;B—蒸汽压力下限温度曲线。
▲图3-不同蒸汽压力条件下产品的温度 - 时间曲线
将测试数据进行对比分析,具体如表 1 所示。①蒸汽压力上限条件下运行升温的时间与日常运行的升温时间基本一致,蒸汽压力下限条件下运行升温的时间比日常运行的升温时间增加 14.67 min。②在蒸汽压力上限和下限条件下运行,灭菌器内温度的均匀性及控温能力均符合规定,但在上限条件下运行,温度的均匀性较日常运行有所降低。③在蒸汽压力上限条件下运行,F0 值与日常运行的 F0 值接近,但在蒸汽压力下限条件下运行,F0 值比日常运行时增加 3.37。
▲表1-温度测试数据的对比分析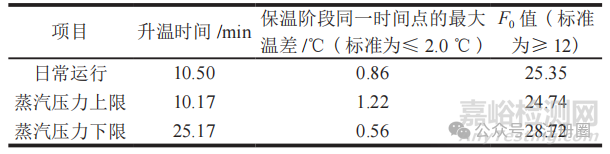
通过上述研究,企业可以了解所使用的灭菌器在不同蒸汽压力下的温度变化情况,以及蒸汽压力对灭菌器的具体影响程度。蒸汽压力过高会对温度均匀性产生一定影响 ;蒸汽压力过低会延长灭菌时间,增加 F0 值。因此,日常生产过程中应尽可能保证蒸汽供应的稳定性,使蒸汽压力在较小范围内波动,以确保能持续、稳定地赋予被灭菌产品所要求的杀灭效果。
2.5 二次污染
应评价现有灭菌系统是否会造成灭菌后产品的二次污染,对可能造成灭菌后产品二次污染的因素进行风险评估,并制定相应措施降低或消除风险。例如,进入灭菌器腔室内的气体 ( 通常是空气或氮气 ) 应有除菌过滤器。水浴式灭菌器可采用换热器对直接接触产品的循环水进行降温,避免直接通入冷却介质造成产品二次污染。
2.6 产品隔离
为防止已灭菌产品与未灭菌产品发生混淆,可采用以下措施降低产品混淆的风险。①在产品生产线上设置隔离物,将已灭菌产品和未灭菌产品分开放置。②使用双扉灭菌器,将未灭菌产品始终控制在灭菌器的一侧,并从同一侧的门向灭菌器内加载产品。灭菌结束后,产品只能从另一侧门卸载,且只能从卸载一侧直接进入外包装区。③在每一灭菌装载 ( 车、架或盘 ) 上应附上灭菌装载指示物 ( 如化学性灭菌指示剂或综合性化学指示剂 ),区分装载上产品的状态。值得注意的是,附在装载上的化学指示剂只能用于区分装载是否经过灭菌程序,不可用于评价被灭菌产品的 SAL。使用前,需确定指示剂的稳定性及其对温度 - 时间的反应特性 [10]。除以上措施外,还可以使用电子化或自动化系统来降低或消除由于人为差错导致的混淆。
灭菌后产品的风险评估应包括产品管理、数量平衡以及已灭菌产品与未灭菌产品的物理 / 化学隔离。在风险评估中应详细分析可能导致已灭菌产品与未灭菌产品混淆的各种失误,并制定相应预防措施。应定期对风险进行回顾性审核,确保所建议的措施能有效地降低风险,且不会引入新的风险。一旦有不良事件、变更或新的重要信息或数据 ( 如偏差 ) 发生,均须对风险进行复核、更新。
2.7 人员
无论是灭菌工艺的验证还是日常生产监控,相关人员需要了解和掌握湿热灭菌的专业知识,并接受相关专业培训。需有 1 名或 1 名以上灭菌人员在生产现场工作,其应具备判定生产是否符合参数放行要求的能力,可解决灭菌系统运行过程中所出现的问题。
应对灭菌岗位、设备维护部门和质量保证部门审核灭菌记录的关键工作人员的专业能力和资质进行评价,培训经历和工作职责均需有文档记录。
3.结语
参数放行基于良好的 GMP 实践,实施参数放行有利于企业深化对产品工艺的理解和控制,有利于提升企业的质量管理水平和对生产过程的控制能力,从而降低生产过程中的差错、混淆、污染与交叉污染。同时,不依赖无菌检测进行产品放行,可降低产品的库存周期与仓储成本,从而提升产品的市场竞争力。
参考文献
[1] 中国医药质量管理协会.湿热灭菌无菌产品参数放行要求[EB/OL]
https://www.doc88.com/p-70699779637648.html?id=3&s=like.
[2] The United States Pharmacopeial Convention.General chapter <1222> terminally sterilized pharmaceutical release [EB/OL].https://www.drugfuture.com/pharmacopoeia/usp32/pub/data/v32270/usp32nf27s0_c1222.html.
[3] 张启林 , 林晶晶 . 湿热灭菌工艺验证探讨 [J]. 中国医药工业杂志 , 2021, 52(8): 1112.
[4] Parenteral Drug Association.Technical report No.48, moistheat sterilizer systems: design, commissioning, operation, qualification, and maintenance [EB/OL].https://pda.org/docs/default-source/website-document-library/chapters/presentations/new-england/pda-technical-report-48-moistheat-sterilizer-systems.pdf?sfvrsn=6.
[5] ECA Academy.EU GMP Annex 17: real time release testing and parametric release [EB/OL].https://www.gmp-compliance.org/guidelines/gmp-guideline/eu-gmp-annex-17-real-time release-testing-and-parametric-release.
[6] Parenteral Drug Association.Technical report No.1(revised2007), validation of moist heat sterilization processes cycle design, development, qualification and ongoing control [EB/OL].https://www.pda.org/bookstore/product-detail/4880-tr-1-validation-of-moist-heat-sterilization.
[7] 国家药品监督管理局食品药品审核查验中心.药品 GMP指南 : 无菌制剂 [M].第 2 版.北京 : 中国医药科技出版社 , 2023: 675-685.
[8] Parenteral Drug Association.Technical report No.30(revised 2012), parametric release of pharmaceutical and medical device products terminally sterilized by moist heat [EB/OL].https://www.pda.org/bookstore/product-detail/1188-tr-30-parametric-releaseof-pharmaceuticals.
[9] 尚 悦 . 无菌药品参数放行国际实施历程及我国现状浅析[J]. 中国医药工业杂志 , 2021, 52(9): 1248-1252.
[10] 潘友文 , 邓海根.灭菌工艺的基本原理与参数放行 [M].北京 : 中国标准出版社 , 2013: 155-618.

来源:中国医药工业杂志
关键词: 灭菌系统