
嘉峪检测网 2025-05-19 09:13
导读:那么LBPs产品非临床研究究竟怎么开展呢?我们结合一个FDA和EMA双报的案例看一下。
活的微生物用于医学应用已经比较常见,这类产品FDA和Ph.Eur《欧洲药典》将其归类为活生物制品(Live Biotherapeutic Products, LBPs)。FDA将这类产品的定义为生物制品,包括3个特点:1)含有活的微生物如细菌;2)用于预防、治疗或治愈一种人类疾病;3)不是疫苗。Ph.Eur的定义则相对简单,用于人体的含有活的微生物(细菌或酵母)的药物,粪便移植微生物或基因治疗产品不包括在内。LBPs活的特点及作用机制的复杂性为有效性和安全性的评估带来很多挑战。
栖息在人体内的复杂微生物生态系统由细菌、古菌、真菌、原生动物和病毒组成,统称为“微生物群”。LBPs既不是通过到达远端器官、组织或受体来发挥其生物效应,也不会直接作用于已知的靶点。那么LBPs是怎么发挥作用的呢?一般认为LBPs是通过调节宿主微生物群发挥作用,例如通过抑制病原体,产生活性分子/代谢物,调节粘膜免疫系统活性,激活上皮细胞内的细胞通路,或调节神经系统的活动。以上作用可全部或部分同时发生。还有一点很重要,LBPs能影响局部生态系统中的其他微生物与宿主的相互作用。此外,微生物群受环境(如压力、营养或药物等)影响很大,且微生物群具有宿主特异性。
监管机构首次提到LBPs是在欧洲药典委员会2019年发布的3053E General monograph on Live Biotherapeutic Products中。不过,里面更多的是讨论对这类产品质量的要求。实际上,目前并没有专门针对LBPs非临床研究的指导原则。
由于微生物群组与宿主的共生关系高度复杂,并且具备高度的种属特异性和依赖性,微生物群的任何差异都可能对宿主的生理学产生重大影响。这就使得动物有效性和安全性研究结果的人体相关性和转化性面临很大挑战。为解决这一问题,建议采用多种不同的方法表征LBP产品的安全性。
为了阐明LBP产品的MOA,通常会用一些体外的工具,如器官芯片、微流控技术等均可以考虑。如果LBP是局部皮肤外用,也有一些3D-皮肤模型或皮肤器官芯片技术可用。体内工具则分为简单动物模型和哺乳动物模型。简单动物模型包括果蝇或秀丽隐杆线虫等。这类模型在评估LBP与宿主之间相互作用机制方面具备优势。哺乳动物在LBP评价方面的缺点主要是人体相关性和临床转化效果比较差。毕竟,动物与人体细菌之间的相互作用和共同进化,与人体是有很大差异的。人体细菌移植到动物体内后,会逐渐向动物宿主细菌成分转化,虽然不会完全转变为动物细菌,但最终会成为一个人体菌和动物菌的混合菌。
LBPs几乎不会进入外周循环,故传统的吸收、分布、代谢等试验并不是必须的。但是,需要了解LBPs给药后,在新的环境中的分布、存活率、转运、生长、表型等。
那么LBPs产品非临床研究究竟怎么开展呢?我们结合一个FDA和EMA双报的案例看一下。
小克里斯滕森氏菌(Christensenella minuta)是一种革兰氏阴性、不产生孢子、不运动的细菌:属于厚壁菌门。法国YSOPIA公司开发了Christensenella minuta DSM 33407,DP名为Xla1,拟用于治疗肥胖和代谢相关疾病。基于其质量、有效性和安全性数据分别于2019年和2020年与FDA和EMA开了pre-IND会议。
CMC
LBP开发起始于细菌菌株的分离、建库和表征。这点类似于传统生物药物的建库要求,建立RCB、MCB和WCB三级库,后面两级库需要在GMP条件下建立,且需要充分表征和记录。需要明确记录菌株来源、菌株培养和传代。如果菌株从人源生物材料分离,则人源供体的信息也要记录,包括年龄、性别、一般生理状况、健康状况或病史、体重指数、无病原体以及采样前一定时期的抗菌治疗。
美国和欧洲对于菌株的表征主要从两个方面考量,一个是基因型,一个是表型。基因型表征内容包括种属鉴别、菌株鉴别、细菌耐药基因、毒力基因、可移动遗传元件、质粒、噬菌体相关DNA、转座子等。表型表征的内容则包括菌株鉴别、形态学鉴定、革兰氏染色、细胞形态和大小、生长特征、运动和孢子形成、抗生素敏感性分析、酶活、细菌内毒素等。有些检测指标如种属鉴别、菌株鉴别,FDA甚至要求采用两种方法交叉验证。
建库之后下一步就是工艺开发。细菌培养是LBPs产品的关键步骤,故需要对工艺过程进行质控。EMA强烈建议就临床Ⅰ期样品制备过程中的关键步骤进行过程控制,并建立接受标准。作为活的微生物,每种细菌尤其特定的生长需求,如培养基和培养参数需要紧密控制。比如,厌氧菌不能通入氧气。一些起始物料也必须按照GMP要求执行。
为降低终产品机会性交叉感染风险,开展微生物和菌株鉴别是确保LBPs产品质量和安全性的关键步骤。微生物检查包括需氧微生物污染计数(AMCC)、酵母菌/霉菌混合污染计数(YMCC)和特定微生物的测试,如16S rDNA基因分型检测。欧洲和美国药典对LBPs的限度和检测方法均有规定。
LBPs的批间差异比其他类型药物要更大,而且终产品的稳定性也不如其他药物。FDA和EMA对这点都表示理解,未提出太多异议。
药理毒理
Christensenella minuta DSM 33407开展的非临床研究如下表所示。与传统药物相比,非临床研究内容简单很多,主要原因是1)Christensenella minuta DSM 33407是一种人体共生菌,已有大量临床数据;2)没有任何动物模型可以准确模拟该菌株在人体胃肠道中的作用;3)为了尽可能重现人体微生物生态,构建一个ex vivo GI SHIME模型。GI SHIME是什么?其实就是因为这类产品没有合适的动物模型,采用的一种人体肠道微生物生态系统模拟器。GI SHIME模型,全称为Simulator of the Human Intestinal Microbial Ecosystem(人类肠道微生物生态系统模拟器),是一种高度复杂的动态模拟系统,用于模拟人体胃肠道的生理、化学和微生物特性。该模型由五个主要部分组成:胃、小肠以及近端、横段和远端结肠,能够模拟食物从摄入到消化、吸收和发酵的全过程。SHIME模型的开发始于1993年,它不仅能够模拟肠道微生物群落的组成和代谢活动,还能模拟肠道内环境参数的变化,如pH、氧化还原电位、营养物质的可用性等。模型的运作需要37℃的恒温条件,通过蠕动泵连接的双层夹套玻璃容器,模拟食物在胃肠道中的流动。每个阶段的pH值都会根据胃肠道的实际条件进行调节,并通过磁力搅拌器保持混合。这一生态模拟系统在Christensenella minuta DSM 33407的非临床研究中发挥了关键作用。
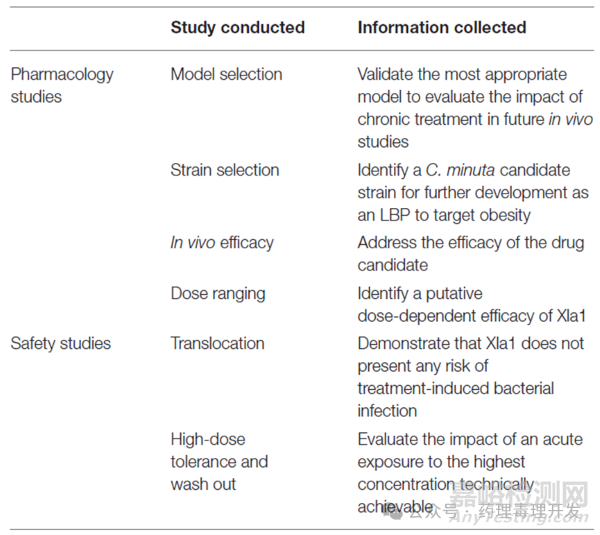
每种细菌的生物学作用各不相同,因此其药理学研究取决于细菌的特点。EMA和FDA均要求根据LBPs临床拟用情况进行非临床研究的设计。不过,如前文所述,LBPs限于临床前模型的缺失,很难实现这一目的。EMA建议先想办法证明LBPs给药后,是否引起病理生理学变化,以及MOA是什么。虽然LBPs的作用机制通常不是单一的,而是通过影响多种通路发挥作用。至少在进入临床试验之前,先弄清楚LBPs是如何发挥作用的。这也是为什么一些人为构建的器官模型派上了用场,如前面提及的SHIME模型。
药代动力学方面,对于LBPs,通常不会进入系统循环,并到达远端器官。因此,Christensenella minuta DSM 33407未开展传统的PK研究,取而代之的是体外生物转运和宿主-微生物相互作用研究。FDA和EMA也认可传统的PK和TK研究对于LBPs并不适用。不过,要有数据证明受试菌株不能进行系统循环,如细菌移位试验。
毒理研究方面,LBPs主要存在两类风险,一种是抗生素耐药转移到其他细菌的风险,一种是引发机体感染的风险。前者是一种重要的耐药性传播机制,包括转化、接合和转导等。通过这些过程,耐药基因可以从一个细菌转移到另一个细菌,即使它们属于不同的物种。例如,质粒是一种常见的可移动遗传元件,可以携带耐药基因并在细菌间传递。后者则是细菌可能通过移位进入外周血,引发感染。由于缺乏可靠的非临床模型,实现这两个风险的评估很大程度上依赖于文献资料收集。根据LBPs菌株特点、临床目标人群等对可能的获益、风险进行文献检索和综述。
当然,并不是任何毒理研究均不开展。Christensenella minuta DSM 33407开展了细菌移位试验。细菌移位指肠道细菌穿过肠黏膜屏障,进入通常无菌的组织如肠系膜淋巴结以及其他器官的过程,可以用qPCR对相关组织进行检测。
FDA认为开展的毒理研究支持Xla1的临床Ⅰ期试验,现阶段不需要开展额外的毒理试验。当然,FDA同样提示,如果本品在临床开发过程中出现一些安全性风险的信号,监管机构将会要求额外的毒理试验进行支持。EMA则未给出清晰结论,只是反馈如果来源于健康的人类共生细菌,其摄入量在生理范围内,不能进入外周系统循环,则不需要开展额外毒理研究。
关于GLP依从性,大部分毒理研究尽可能在GLP条件下开展。但是,一些厌氧菌需要特定的研究条件,很多GLP机构不具备开展条件。这种情况下,监管机构接受non-GLP条件下产生的毒理数据。但是,研究质量并不能打折扣。而且,EMA额外强调,应该讨论偏离GLP条件可能产生的影响。

来源:药理毒理开发
关键词: 生物制品非临床研究